Terahertz Time Domain Spectroscopy of Amino Acids and Sugars
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Application of the Theory of Laser to Nitrogen Laser Pumped Dye Laser
SD9900039 AN APPLICATION OF THE THEORY OF LASER TO NITROGEN LASER PUMPED DYE LASER FATIMA AHMED OSMAN A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Physics. UNIVERSITY OF KHARTOUM FACULTY OF SCIENCE DEPARTMENT OF PHYSICS MARCH 1998 \ 3 0-44 In this thesis we gave a general discussion on lasers, reviewing some of are properties, types and applications. We also conducted an experiment where we obtained a dye laser pumped by nitrogen laser with a wave length of 337.1 nm and a power of 5 Mw. It was noticed that the produced radiation possesses ^ characteristic^ different from those of other types of laser. This' characteristics determine^ the tunability i.e. the possibility of choosing the appropriately required wave-length of radiation for various applications. DEDICATION TO MY BELOVED PARENTS AND MY SISTER NADI A ACKNOWLEDGEMENTS I would like to express my deep gratitude to my supervisor Dr. AH El Tahir Sharaf El-Din, for his continuous support and guidance. I am also grateful to Dr. Maui Hammed Shaded, for encouragement, and advice in using the computer. Thanks also go to Ustaz Akram Yousif Ibrahim for helping me while conducting the experimental part of the thesis, and to Ustaz Abaker Ali Abdalla, for advising me in several respects. I also thank my teachers in the Physics Department, of the Faculty of Science, University of Khartoum and my colleagues and co- workers at laser laboratory whose support and encouragement me created the right atmosphere of research for me. Finally I would like to thank my brother Salah Ahmed Osman, Mr. -

High-Power Solid-State Lasers from a Laser Glass Perspective
LLNL-JRNL-464385 High-Power Solid-State Lasers from a Laser Glass Perspective J. H. Campbell, J. S. Hayden, A. J. Marker December 22, 2010 Internationakl Journal of Applied Glass Science Disclaimer This document was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor Lawrence Livermore National Security, LLC, nor any of their employees makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or Lawrence Livermore National Security, LLC. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or Lawrence Livermore National Security, LLC, and shall not be used for advertising or product endorsement purposes. High-Power Solid-State Lasers from a Laser Glass Perspective John H. Campbell, Lawrence Livermore National Laboratory, Livermore, CA Joseph S. Hayden and Alex Marker, Schott North America, Inc., Duryea, PA Abstract Advances in laser glass compositions and manufacturing have enabled a new class of high-energy/high- power (HEHP), petawatt (PW) and high-average-power (HAP) laser systems that are being used for fusion energy ignition demonstration, fundamental physics research and materials processing, respectively. The requirements for these three laser systems are different necessitating different glasses or groups of glasses. -

Simultaneous Intensity Profiling of Multiple Laser Beams Using the Bladecam-XHR Camera
Simultaneous Intensity Profiling of Multiple Laser Beams Using the BladeCam-XHR Camera Shaun Pacheco College of Optical Sciences, University of Arizona Rongguang Liang College of Optical Sciences, University of Arizona Rocco Dragone Vice President of Engineering, DataRay, Inc. Real-Time Profiling of Multiple Laser Beams There are several applications where the parallel processing of multiple beams can significantly decrease the overall time needed for the process. Intensity profile measurements that can characterize each of those beams can lead to improvements in that application. If each beam had to be characterized individually, the process would be very time consuming, especially for large numbers of beams. This white paper describes how the BladeCam-XHR can be used to simultaneously measure the intensity profile measurements for multiple beams by measuring the diffraction pattern from a diffraction grating and the intensity profile from a 3x3 fiber array focused using a 0.5 NA objective. Diffraction Gratings Diffraction gratings are periodic optical components that diffract an incident beam into several outgoing beams. The grating profile determines both the angle of the diffracted beam modes and the efficiency of light sent into each of those modes. Grating fabrication errors or a change in the incident wavelength can significantly change the performance of a diffraction grating. The BladeCam- XHR, Figure 1, can be used to measure the point spread function (PSF) of each mode, the relative efficiency of each mode and the spacing between diffraction modes. Figure 1 BladeCam-XHR Using the BladeCam-XHR for Diffraction Grating Evaluation The profiling mode in the DataRay software can be used to evaluate the performance of a diffraction grating. -

Applications of Laser Spectroscopy Constitute a Vast Field, Which Is Difficult to Spectroscopy Cover Comprehensively in a Review
LASERS & OPTICS 151 many cases non-destructively. A variety of beam manipulation schemes are avail able for the irradiation of samples either directly or after preparation. The unique properties of laser radiation in terms of coherence, intensity and directionality permit remote chemical sensing, where the analytical equipment and the sample are separated by large distances, even of several kilometres. Absorption, and in particular differential absorption, can be utilised in long-path measurements, whereas elastic and inelastic backscatter- ing as well as fluorescence can be used for range-resolved radar-like measure ments (LIDAR: light detection and rang ing). Laser light can also be efficiently transported in optical fibres to remotely located measurement sites. Various prop erties of the fibre itself influence the laser light propagating through the fibre, thus Applications of Laser forming a basis for fibre optic sensors. Applications of laser spectroscopy constitute a vast field, which is difficult to Spectroscopy cover comprehensively in a review. Rather than attempting such a review, examples from a variety of fields are cho Costas Fotakis sen for illustrating the power of applied University of Crete, Greece laser spectroscopy. Sune Svanberg Lund Institute of Technology, Sweden Applications in Analytical Chemistry Laser spectroscopy is making a major impact in many traditional fields of ana As the new millennium approaches the Above The Italian research vessel Urania, carrying a lytical spectroscopy. For instance, opto- know how in the field of laser-matter Swedish laser radar system, which has sailed under the galvanic spectroscopy on analytical smoky plumes of Sicilian volcanos in Italy measuring the interactions has matured to the stage of sulphur dioxide content flames increases the sensitivity of enabling several exciting real life applica absorption and emission flame spec tions. -

Hair Reduction by Laser
Ames Locations To Eldora, Iowa Falls, McFarland Clinic North Ames Story City, Urgent Care Webster City Treatment McFarland Clinic (3815 Stange Rd.) Bloomington Road McFarland Clinic Physical Therapy - Somerset West Ames e. (2707 Stange Rd., Suite 102) 24th Street Av f e. Duf Av You should not use any Accutane (a prescribed oral US 69 Ontario Street 13th Street Grand 13th Street acne medication) or gold medications (for arthritis) McFarland Clinic (1215 Duff) MGMC - North Addition for six months prior to a treatment. You should William R. Bliss Cancer Center 12th Street Stange Road . e. Mary Greeley Medical Center (MGMC) McFarland Eye Center not use retinoids (e.g. Retin A, Fifferin, Differin, Av (1111 Duff Ave.) (1128 Duff) e. 11th Street Ramp Av McFarland Family Avita, Tazorac), alpha-hydroxy acid moisturizers, Medicine East Hyland St Medical Arts Building (1018 Duff) To Boone, Carroll I-35 bleaching creams (hydroquinone) or chemical peels Iowa State (1015 Duff) Jefferson N. Dakota Marshall University for two weeks prior to a treatment. Do not pluck the Lincoln Way Lincoln Way Express Care Express Care hairs, wax or use depilatories or electrolysis for six e. (3800 Lincoln Way) (640 Lincoln Way) weeks prior to a treatment. Av McFarland Clinic West Ames Hy-Vee (3600 Lincoln Way) Hy-Vee e. Av ff You may be asked to shave the area prior to your S. Dakota To Nevada, Du first treatment appointment. You may also be Marshalltown prescribed a topical anesthetic (numbing) cream to Highway 30 help reduce any discomfort the laser may cause. Blvd University First Treatment: The laser will be used to treat the N areas by your dermatologist or one of the trained Oakwood Road Airport Road dermatology staff. -

Laser Pointer Safety About Laser Pointers Laser Pointers Are Completely Safe When Properly Used As a Visual Or Instructional Aid
Harvard University Radiation Safety Services Laser Pointer Safety About Laser Pointers Laser pointers are completely safe when properly used as a visual or instructional aid. However, they can cause serious eye damage when used improperly. There have been enough documented injuries from laser pointers to trigger a warning from the Food and Drug Administration (Alerts and Safety Notifications). Before deciding to use a laser pointer, presenters are reminded to consider alternate methods of calling attention to specific items. Most presentation software packages offer screen pointer options with the same features, but without the laser hazard. Selecting a Safe Laser Pointer 1) Choose low power lasers (Class 2) Whenever possible, select a Class 2 laser pointer because of the lower risk of eye damage. 2) Choose red-orange lasers (633 to 650 nm wavelength, choose closer to 635 nm) The National Institute of Standards and Technology (NIST) researchers found that some green laser pointers can emit harmful levels of infrared radiation. The green laser pointers create green light beam in a three step process. A standard laser diode first generates near infrared light with a wavelength of 808nm, and pumps it into a ND:YVO4 crystal that converts the 808 nm light into infrared with a wavelength of 1064 nm. The 1064 nm light passes into a frequency doubling crystal that emits green light at a wavelength of 532nm finally. In most units, a combination of coatings and filters keeps all the infrared energy confined. But the researchers found that really inexpensive green lasers can lack an infrared filter altogether. One tested unit was so flawed that it released nine times more infrared energy than green light. -

Population Inversion X-Ray Laser Oscillator
Population inversion X-ray laser oscillator Aliaksei Halavanaua, Andrei Benediktovitchb, Alberto A. Lutmanc , Daniel DePonted, Daniele Coccoe , Nina Rohringerb,f, Uwe Bergmanng , and Claudio Pellegrinia,1 aAccelerator Research Division, SLAC National Accelerator Laboratory, Menlo Park, CA 94025; bCenter for Free Electron Laser Science, Deutsches Elektronen-Synchrotron, Hamburg 22607, Germany; cLinac & FEL division, SLAC National Accelerator Laboratory, Menlo Park, CA 94025; dLinac Coherent Light Source, SLAC National Accelerator Laboratory, Menlo Park, CA 94025; eLawrence Berkeley National Laboratory, Berkeley, CA 94720; fDepartment of Physics, Universitat¨ Hamburg, Hamburg 20355, Germany; and gStanford PULSE Institute, SLAC National Accelerator Laboratory, Menlo Park, CA 94025 Contributed by Claudio Pellegrini, May 13, 2020 (sent for review March 23, 2020; reviewed by Roger Falcone and Szymon Suckewer) Oscillators are at the heart of optical lasers, providing stable, X-ray free-electron lasers (XFELs), first proposed in 1992 transform-limited pulses. Until now, laser oscillators have been (8, 9) and developed from the late 1990s to today (10), are a rev- available only in the infrared to visible and near-ultraviolet (UV) olutionary tool to explore matter at the atomic length and time spectral region. In this paper, we present a study of an oscilla- scale, with high peak power, transverse coherence, femtosecond tor operating in the 5- to 12-keV photon-energy range. We show pulse duration, and nanometer to angstrom wavelength range, that, using the Kα1 line of transition metal compounds as the but with limited longitudinal coherence and a photon energy gain medium, an X-ray free-electron laser as a periodic pump, and spread of the order of 0.1% (11). -

Fundamentals of Frequency Combs: What They Are and How They Work
Fundamentals of frequency combs: What they are and how they work Scott Diddams Time and Frequency Division National Institute of Standards and Technology Boulder, CO KISS Worshop: “Optical Frequency Combs for Space Applications” 1 Outline 1. Optical frequency combs in timekeeping • How we got to where we are now…. 2. The multiple faces of an optical frequency comb 3. Classes of frequency combs and their basic operation principles • Mode-locked lasers • Microcombs • Electro-optic frequency combs 4. Challenges and opportunities for frequency combs 2 Timekeeping: The long view Oscillator Freq. f = 1 Hz Rate of improvement f = 10 kHz since 1950: 1.2 decades / 10 years f = 10 GHz “Moore’s Law”: 1.5 decades / 10 years f = 500 THz Gravitational red shift of 10 cm Sr optical clock ? f = 10-100 PHz ?? S. Diddams, et al, Science (2004) Timekeeping: The long view Oscillator Freq. f = 1 Hz f = 10 kHz f = 10 GHz f = 500 THz Gravitational red shift of 10 cm Sr optical clock ? f = 10-100 PHz ?? S. Diddams, et al, Science (2004) Use higher frequencies ! Dividing the second into smaller pieces yields superior frequency standards and metrology tools: 15 νoptical 10 ≈ ≈ 105 νmicrowave 1010 In principle (!), optical standards could surpass their microwave counterparts by up to five orders of magnitude ... but how can one count, control and measure optical frequencies? 5 Optical Atomic Clocks Atom(s) νo Detector At NIST/JILA: Ca, Hg+, Al+, Yb, Sr & Slow Δν Laser Control Atomic Resonance Counter & Laser Read Out Fast Laser (laser frequency comb) Control •Isolated cavity narrows laser linewidth and provides short term timing reference Isolated Optical Cavity •Atoms provide long-term stability and accuracy— now now at 10-18 level! •Laser frequency comb acts as a divider/counter Outline 1. -
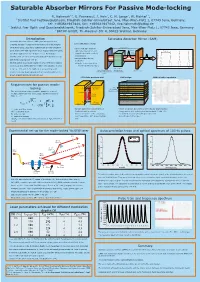
Saturable Absorber Mirrors for Passive Mode-Locking
Saturable Absorber Mirrors For Passive Mode-locking R. Hohmuth1 3, G. Paunescu2, J. Hein2, C. H. Lange3, W. Richter1 3, 1 Institut fuer Festkoerperphysik, Friedrich-Schiller-Universitaet Jena, Max-Wien-Platz 1, 07743 Jena, Germany, tel: +493641947444, fax: +493641947442, [email protected] 2 Institut fuer Optik und Quantenelektronik, Friedrich-Schiller-Universitaet Jena, Max-Wien-Platz 1, 07743 Jena, Germany 3 BATOP GmbH, Th.-Koerner-Str. 4, 99425 Weimar, Germany Introduction Saturable Absorber Mirror (SAM) Saturable absorber mirrors (SAMs) are inexpensive and schematic laser set-up compact devices for passive mode-locking of diode pumped solid state lasers. Such laser systems can provide ultrashort cavity, length L -cavity with gain medium pulse trains with high repetition rates. Typical values for pulse pulse -high reflective mirror and TRT=2L/c ) t output mirror with partially ( duration ranging from 100 fs up to 10 ps. For instance a I y t transmission i s Nd:YAG laser can be mode-locked with pulse duration of 8 ps n e -saturable absorber as t n and mean output power of 6 W. I modulator On this poster we present results for a Yb: KYW laser passive =>pulse trains spaced by mode-locked by SAMs with three different modulation depths round-trip time TRT=2L/c time t between 0.6% and 2.0%. SAMs were prepared by solid- gain medium high reflective mirror (pumped e.g. output mirror source molecular beam epitaxy with a low-temperature (LT) with saturable absorber by laser diode) (SAM) grown InGaAs absorbing quantum well. LT InGaAs quantum well SAM design SAM reflection spectrum Ta O or SiO / dielectric cover Requirements for passive mode- 25 2 conduction 71.2 nm GaAs / barrier E 7 nm c1 71.2 nm GaAs / barrier locking band Ec LT InGaAs 74.7 nm GaAs The mode-locking regime is stable agaist the onset of quantum well InGaAs 88.4 nm AlAs multiple pulsing as long as the pulse duration is smaller energy : 25x Bragg mirror than t . -

Towards On-Chip Self-Referenced Frequency-Comb Sources Based on Semiconductor Mode-Locked Lasers
micromachines Review Towards On-Chip Self-Referenced Frequency-Comb Sources Based on Semiconductor Mode-Locked Lasers Marcin Malinowski 1,*, Ricardo Bustos-Ramirez 1 , Jean-Etienne Tremblay 2, Guillermo F. Camacho-Gonzalez 1, Ming C. Wu 2 , Peter J. Delfyett 1,3 and Sasan Fathpour 1,3,* 1 CREOL, The College of Optics and Photonics, University of Central Florida, Orlando, FL 32816, USA; [email protected] (R.B.-R.); [email protected] (G.F.C.-G.); [email protected] (P.J.D.) 2 Department of Electrical Engineering and Computer Sciences, University of California, Berkeley, CA 94720, USA; [email protected] (J.-E.T.); [email protected] (M.C.W.) 3 Department of Electrical and Computer Engineering, University of Central Florida, Orlando, FL 32816, USA * Correspondence: [email protected] (M.M.); [email protected] (S.F.) Received: 14 May 2019; Accepted: 5 June 2019; Published: 11 June 2019 Abstract: Miniaturization of frequency-comb sources could open a host of potential applications in spectroscopy, biomedical monitoring, astronomy, microwave signal generation, and distribution of precise time or frequency across networks. This review article places emphasis on an architecture with a semiconductor mode-locked laser at the heart of the system and subsequent supercontinuum generation and carrier-envelope offset detection and stabilization in nonlinear integrated optics. Keywords: frequency combs; heterogeneous integration; second-harmonic generation; supercontinuum; integrated photonics; silicon photonics; mode-locked lasers; nonlinear optics 1. Introduction The field of integrated photonics aims at harnessing optical waves in submicron-scale devices and circuits, for applications such as transmitting information (communications) and gathering information about the environment (imaging, spectroscopy, etc.). -

Early-Stage Dynamics of Chloride Ion–Pumping Rhodopsin Revealed by a Femtosecond X-Ray Laser
Early-stage dynamics of chloride ion–pumping rhodopsin revealed by a femtosecond X-ray laser Ji-Hye Yuna,1, Xuanxuan Lib,c,1, Jianing Yued, Jae-Hyun Parka, Zeyu Jina, Chufeng Lie, Hao Hue, Yingchen Shib,c, Suraj Pandeyf, Sergio Carbajog, Sébastien Boutetg, Mark S. Hunterg, Mengning Liangg, Raymond G. Sierrag, Thomas J. Laneg, Liang Zhoud, Uwe Weierstalle, Nadia A. Zatsepine,h, Mio Ohkii, Jeremy R. H. Tamei, Sam-Yong Parki, John C. H. Spencee, Wenkai Zhangd, Marius Schmidtf,2, Weontae Leea,2, and Haiguang Liub,d,2 aDepartment of Biochemistry, College of Life Sciences and Biotechnology, Yonsei University, Seodaemun-gu, 120-749 Seoul, South Korea; bComplex Systems Division, Beijing Computational Science Research Center, Haidian, 100193 Beijing, People’s Republic of China; cDepartment of Engineering Physics, Tsinghua University, 100086 Beijing, People’s Republic of China; dDepartment of Physics, Beijing Normal University, Haidian, 100875 Beijing, People’s Republic of China; eDepartment of Physics, Arizona State University, Tempe, AZ 85287; fPhysics Department, University of Wisconsin, Milwaukee, Milwaukee, WI 53201; gLinac Coherent Light Source, Stanford Linear Accelerator Center National Accelerator Laboratory, Menlo Park, CA 94025; hDepartment of Chemistry and Physics, Australian Research Council Centre of Excellence in Advanced Molecular Imaging, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC 3086, Australia; and iDrug Design Laboratory, Graduate School of Medical Life Science, Yokohama City University, 230-0045 Yokohama, Japan Edited by Nicholas K. Sauter, Lawrence Berkeley National Laboratory, Berkeley, CA, and accepted by Editorial Board Member Axel T. Brunger February 21, 2021 (received for review September 30, 2020) Chloride ion–pumping rhodopsin (ClR) in some marine bacteria uti- an acceptor aspartate (13–15). -

Atomic Clocks of the Future: Using the Ultrafast and Ultrastable'
Atomic clocks of the future: using the ultrafast and ultrastable' Leo Hollberg, Scott Diddanis, Chris Oates, Anne Curtis. SCbastien Bize, and Jim Bergquist National Institute of Standards and Technology, 325 Broadway, Boulder, CO 80305 F-mai!: !~ol!ber~~,b~~!der.ni~~.gc\~ *Contribution of NlST not subject Lo copyrigl~t. Abstract. The applicalion of ultrafast mode-locked lasers and nonlinear optics to optical frequency nietrology is revolutionizing the field ofatomic clocks. The basic concepts and applications are reviewed using our recent rcsults with two optical atomic frequency standards based on laser cooled and trapped atoms. In contrast to today's atomic clocks that are based on electronic oscillators locked to microwave transitions in atoms, the nest generation of atomic clocks will ]nos[ likely employ lasers and optical transitions in lasel--cooled atoms and ions. Optical atomic frequency standards use frequency-stabilized CW lasers with good short-term stability that are stabilized to narrow atomic rcsonances using feedback control systems. At NIST we are developing two optical frequency standards, one based on laser cooled and trapped calcium atoms and the other on a single laser cooled trapped Hg+ ion. Similar research is being done at labs around the world. 1\1oving from microwave frequencies, where one cycle corresponds to 100 ps, to optical frequencies where one cycle is a femtosecond long, allows us to divide time into smaller intervals and hence gain frequency stability and timing precision. Steady progress over the past 30 years has improved the performance of optical fiquency standards to the point that they are now competitive with, and even moving beyond.