Principles of Organic Chemistry Reactive Intermediates Carbocation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reactivity and Mechanism of the Reactions in Volving Carbonyl Ylide
Bull, i Korean Chem. Soc.t Vol. 14, No. 1, 1993 149 10. 0. Inganas, R. Erlandsson, C. Nylander, and I. Lunds- eration of other types of ylides.6 Even though many studies trom, J. Phys. Chem. Solids, 45, 427 (1984). have been reported for the intermediates of carbonyl ylides 11. P. Novak, B. Rasch, and W. Vielstich, J. Electro사 诚m Soc., in the reactions of carbenes with a carbonyl oxygens,7 the 138, 3300 (1991). reactions are limited to the discussion of the transition metal 12. K. M. Cheung, D. Bloor, and G. C. Stevens, Polymers, catalyzed decomposition of a diazo compounds in the prese 29, 1709 (1988). nce of a carbonyl group. In this paper we report the reactivity and mechanism of the reactions involving carbonyl ylide intermediate from the photolytic kinetic results of non-catalytic decompositions of diazomethane and diphenyldiazomethane in the presence of a carbonyl group. Reactivity and Mechanism of the Reactions In volving Carbonyl Ylide Intermediate Experimentals Dae Dong Sung*, Kyu Chui Kim, Sang Hoon Lee, General Photolysis Conditions. Diazomethane was and Zoon Ha Ryu* generated by Aldrich MNNG Diazald apparatus as follows.8 1 mmol of l-methyl-3-nitro-l-nitrosoguanidine (MNNG) is Department of Chemistry, placed in the inside tube of. the Diazald kit through its screw Dong-A University, Pusan 604-714 cap opening along with 0.5 mL of water of dissipate any ^Department of Chemistry, heat generated. 3 mL of diethyl ether is placed in the outside Dong-Eui University, Pusan 614-010 tube and the two parts are assembled with a butyl "O”-ring and held with a pinch-type clamp. -

Free Radical Reactions (Read Chapter 4) A
Chemistry 5.12 Spri ng 2003, Week 3 / Day 2 Handout #7: Lecture 11 Outline IX. Free Radical Reactions (Read Chapter 4) A. Chlorination of Methane (4-2) 1. Mechanism (4-3) B. Review of Thermodynamics (4-4,5) C. Review of Kinetics (4-8,9) D. Reaction-Energy Diagrams (4-10) 1. Thermodynamic Control 2. Kinetic Control 3. Hammond Postulate (4-14) 4. Multi-Step Reactions (4-11) 5. Chlorination of Methane (4-7) Suggested Problems: 4-35–37,40,43 IX. A. Radical Chlorination of Methane H heat (D) or H H C H Cl Cl H C Cl H Cl H light (hv) H H Cl Cl D or hv D or hv etc. H C Cl H C Cl H Cl Cl C Cl H Cl Cl Cl Cl Cl H H H • Why is light or heat necessary? • How fast does it go? • Does it give off or consume heat? • How fast are each of the successive reactions? • Can you control the product ratio? To answer these questions, we need to: 1. Understand the mechanism of the reaction (arrow-pushing!). 2. Use thermodynamics and kinetics to analyze the reaction. 1 1. Mechanism of Radical Chlorination of Methane (Free-Radical Chain Reaction) Free-radical chain reactions have three distinct mechanistic steps: • initiation step: generates reactive intermediate • propagation steps: reactive intermediates react with stable molecules to generate other reactive intermediates (allows chain to continue) • termination step: side-reactions that slow the reaction; usually combination of two reactive intermediates into one stable molecule Initiation Step: Cl2 absorbs energy and the bond is homolytically cleaved. -

Photoionization Studies of Reactive Intermediates Using Synchrotron
Photoionization Studies of Reactive Intermediates using Synchrotron Radiation by John M.Dyke* School of Chemistry University of Southampton SO17 1BJ UK *e-mail: [email protected] 1 Abstract Photoionization of reactive intermediates with synchrotron radiation has reached a sufficiently advanced stage of development that it can now contribute to a number of areas in gas-phase chemistry and physics. These include the detection and spectroscopic study of reactive intermediates produced by bimolecular reactions, photolysis, pyrolysis or discharge sources, and the monitoring of reactive intermediates in situ in environments such as flames. This review summarises advances in the study of reactive intermediates with synchrotron radiation using photoelectron spectroscopy (PES) and constant-ionic-state (CIS) methods with angular resolution, and threshold photoelectron spectroscopy (TPES), taking examples mainly from the recent work of the Southampton group. The aim is to focus on the main information to be obtained from the examples considered. As future research in this area also involves photoelectron-photoion coincidence (PEPICO) and threshold photoelectron-photoion coincidence (TPEPICO) spectroscopy, these methods are also described and previous related work on reactive intermediates with these techniques is summarised. The advantages of using PEPICO and TPEPICO to complement and extend TPES and angularly resolved PES and CIS studies on reactive intermediates are highlighted. 2 1.Introduction This review is organised as follows. After an Introduction to the study of reactive intermediates by photoionization with fixed energy photon sources and synchrotron radiation, a number of Case Studies are presented of the study of reactive intermediates with synchrotron radiation using angle resolved PES and CIS, and TPE spectroscopy. -

Fundamentals of Theoretical Organic Chemistry Lecture 9
Fund. Theor. Org. Chem 1 SE Fundamentals of Theoretical Organic Chemistry Lecture 9 1 Fund. Theor. Org. Chem 2 SE 2.2.2 Electrophilic substitution The reaction which takes place between a reactant with an electronegative carbon and an electropositive reagent forming a polarized covalent bond is called electrophilic. In addition, if substitution occurs (i.e. there is a similar polarized covalent bond on the electronegative carbon, which breaks up during the reaction, so the reagent „substitutes” the „old” group or the leaving group) then this specific reaction is called electrophilic substitution. The electronegative carbon is called the reaction centre. In general, the good reactant are molecules having electronegative carbons like aromatic compounds, alkenes and other compounds containing electron-rich double bonds. These are called Lewis bases. On the contrary, good electrophilic reagents are electron poor compounds/molecule groups like acid-halides, which easily form covalent bond with an electronegative centre, thus creating a new molecule. These are often referred to as Lewis acids. According to molecular orbital (MO) theory the driving force for the electrophilic substitution (SE) is a Lewis complex formation involving the LUMO of the Lewis Acid Reagent and the HOMO of the Lewis Base Reactant. Reactant Reagent LUMO LUMO HOMO Lewis base Lewis acid Lewis complex Types of reactions: There are four types of reactions as illustrated below: 2 Fund. Theor. Org. Chem 3 SE Table. 2.2.2-1. Saturated Aromatic SE1 SE1 (Ar) SE2 SE2 (Ar) SE reaction on saturated atom: (1)Unimolecular electrophilic substitution (SE1): The reaction proceeds in two steps. After the departure of the leaving group, the negatively charged reaction intermediate will combine with the reagent. -

Bsc Chemistry
Subject Chemistry Paper No and Title 05, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) Module No and Title 15, The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds Module Tag CHE_P5_M15 CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. NGP Participation 3.1 NGP by Heteroatom Lone Pairs 3.2 NGP by alkene 3.3 NGP by Cyclopropane, Cyclobutane or a Homoallyl group 3.4 NGP by an Aromatic Ring 4. Neighbouring Group Participation on SN2 Reactions 5. Neighbouring Group Participation on SN1 Reactions 6. Neighbouring Group and Rearrangement 7. Examples 8. Summary CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds 1. Learning Outcomes After studying this module, you shall be able to Know about NGP reaction Learn reaction mechanism of NGP reaction Identify stereochemistry of NGP reaction Evaluate the factors affecting the NGP reaction Analyse Phenonium ion, NGP by alkene, and NGP by heteroatom. 2. Introduction The reaction centre (carbenium centre) has direct interaction with a lone pair of electrons of an atom or with the electrons of s- or p-bond present within the parent molecule but these are not in conjugation with the reaction centre. A distinction is sometimes made between n, s and p- participation. An increase in rate due to Neighbouring Group Participation (NGP) is known as "anchimeric assistance". "Synartetic acceleration" happens to be the special case of anchimeric assistance and applies to participation by electrons binding a substituent to a carbon atom in a β- position relative to the leaving group attached to the α-carbon atom. -

Course Material 2.Pdf
Reactive Intermediates Source: https://www.askiitians.com/iit-jee-chemistry/organic-chemistry/iupac- and-goc/reaction-intermediates/ Table of Content • Carbocations • Carbanions • Free Radicals • Carbenes • Arenium Ions • Benzynes Synthetic intermediate are stable products which are prepared, isolated and purified and subsequently used as starting materials in a synthetic sequence. Reactive intermediate, on the other hand, are short lived and their importance lies in the assignment of reaction mechanisms on the pathway from the starting substrate to stable products. These reactive intermediates are not isolated, but are detected by spectroscopic methods, or trapped chemically or their presence is confirmed by indirect evidence. • Carbocations Carbocations are the key intermediates in several reactions and particularly in nucleophilic substitution reactions. Structure of Carbocations : Generally, in the carbocations the positively charged carbon atom is bonded to three other atoms and has no nonbonding electrons. It is sp 2 hybridized with a planar structure and bond angles of about 120°. There is a + vacant unhybridized p orbital which in the case of CH 3 lies perpendicular to the plane of C—H bonds. Stability of Carbocations: There is an increase in carbocation stability with additional alkyl substitution. Thus one finds that addition of HX to three typical olefins decreases in the order (CH 3)2C=CH 2>CH 3—CH = CH 2 > CH 2 = CH 2. This is due to the relative stabilities of the carbocations formed in the rate determining step which in turn follows from the fact that the stability is increased by the electron releasing methyl group (+I), three such groups being more effective than two, and two more effective than one. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Trapping and Spectroscopic Characterization of an Fe -Superoxo
Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme Michael M. Mbughunia, Mrinmoy Chakrabartib, Joshua A. Haydenb, Emile L. Bominaarb, Michael P. Hendrichb, Eckard Münckb, and John D. Lipscomba,1 aDepartment of Biochemistry, Molecular Biology and Biophysics, and Center for Metals in Biocatalysis, University of Minnesota, Minneapolis, MN 55455, and bDepartment of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213 Edited by Edward I. Solomon, Stanford University, Stanford, CA, and approved August 4, 2010 (received for review July 9, 2010) III •− Fe -O2 intermediates are well known in heme enzymes, but none have been characterized in the nonheme mononuclear FeII enzyme family. Many steps in the O2 activation and reaction cycle of FeII-containing homoprotocatechuate 2,3-dioxygenase are made detectable by using the alternative substrate 4-nitrocatechol (4NC) and mutation of the active site His200 to Asn (H200N). Here, the first intermediate (Int-1) observed after adding O2 to the H200N- 4NC complex is trapped and characterized using EPR and Möss- ∕ III bauer (MB) spectroscopies. Int-1 is a high-spin (S1 ¼ 5 2) Fe anti- ∕ ≈ −1 ferromagnetically (AF) coupled to an S2 ¼ 1 2 radical (J 6cm in H • ¼ JS1 S2). It exhibits parallel-mode EPR signals at g ¼ 8.17 from the S ¼ 2 multiplet, and g ¼ 8.8 and 11.6 from the S ¼ 3 multiplet. 17 These signals are broadened significantly by O2 hyperfine inter- ≈ III •− actions (A17O 180 MHz). Thus, Int-1 is an AF-coupled Fe -O2 spe- cies. The experimental observations are supported by density functional theory calculations that show nearly complete transfer of spin density to the bound O2. -

Lecture Outline a Closer Look at Carbocation Stabilities and SN1/E1 Reaction Rates We Learned That an Important Factor in the SN
Lecture outline A closer look at carbocation stabilities and SN1/E1 reaction rates We learned that an important factor in the SN1/E1 reaction pathway is the stability of the carbocation intermediate. The more stable the carbocation, the faster the reaction. But how do we know anything about the stabilities of carbocations? After all, they are very unstable, high-energy species that can't be observed directly under ordinary reaction conditions. In protic solvents, carbocations typically survive only for picoseconds (1 ps - 10–12s). Much of what we know (or assume we know) about carbocation stabilities comes from measurements of the rates of reactions that form them, like the ionization of an alkyl halide in a polar solvent. The key to connecting reaction rates (kinetics) with the stability of high-energy intermediates (thermodynamics) is the Hammond postulate. The Hammond postulate says that if two consecutive structures on a reaction coordinate are similar in energy, they are also similar in structure. For example, in the SN1/E1 reactions, we know that the carbocation intermediate is much higher in energy than the alkyl halide reactant, so the transition state for the ionization step must lie closer in energy to the carbocation than the alkyl halide. The Hammond postulate says that the transition state should therefore resemble the carbocation in structure. So factors that stabilize the carbocation also stabilize the transition state leading to it. Here are two examples of rxn coordinate diagrams for the ionization step that are reasonable and unreasonable according to the Hammond postulate. We use the Hammond postulate frequently in making assumptions about the nature of transition states and how reaction rates should be influenced by structural features in the reactants or products. -

Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and Its Interactions with Supramolecular Systems
View metadata,Downloaded citation and from similar orbit.dtu.dk papers on:at core.ac.uk May 03, 2019 brought to you by CORE provided by Online Research Database In Technology Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Reynisson, Johannes Publication date: 2000 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Reynisson, J. (2000). Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems. Roskilde: Risø National Laboratory. Denmark. Forskningscenter Risoe. Risoe-R, No. 1191(EN) General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Jóhannes Reynisson OO O Risø National Laboratory, Roskilde, Denmark June 2000 1 Abstract Trioxatriangulenium (TOTA+, 4,8,12-trioxa-4,8,12,12c-tetrahydro- dibenzo[cd,mn]-pyrenylium) is a closed shell carbocation which is stable in its crystalline form and in polar solvents at ambient temperatures. -

Nonclassical Carbocations: from Controversy to Convention
Nonclassical Carbocations H H C From Controversy to Convention H H H A Stoltz Group Literature Meeting brought to you by Chris Gilmore June 26, 2006 8 PM 147 Noyes Outline 1. Introduction 2. The Nonclassical Carbocation Controversy - Winstein, Brown, and the Great Debate - George Olah and ending the discussion - Important nonclassical carbocations 3. The Nature of the Nonclassical Carbocation - The 3-center, 2-electron bond - Cleaving C-C and C-H σ−bonds - Intermediate or Transition state? Changing the way we think about carbocations 4. Carbocations, nonclassical intermediates, and synthetic chemistry - Biosynthetic Pathways - Steroids, by W.S. Johnson - Corey's foray into carbocationic cascades - Interesting rearrangements - Overman and the Prins-Pinacol Carbocations: An Introduction Traditional carbocation is a low-valent, trisubstituted electron-deficient carbon center: R superacid R R LG R R R R R R "carbenium LGH ion" - 6 valence e- - planar structure - empty p orbital Modes of stabilization: Heteroatomic Assistance π-bond Resonance σ-bond Participation R X H2C X Allylic Lone Pair Anchimeric Homoconjugation Hyperconjugation Non-Classical Resonance Assistance Stabilization Interaction Outline 1. Introduction 2. The Nonclassical Carbocation Controversy - Winstein, Brown, and the Great Debate - George Olah and ending the discussion - Important nonclassical carbocations 3. The Nature of the Nonclassical Carbocation - The 3-center, 2-electron bond - Cleaving C-C and C-H σ−bonds - Intermediate or Transition state? Changing the way we think about carbocations 4. Carbocations, nonclassical intermediates, and synthetic chemistry - Biosynthetic Pathways - Steroids, by W.S. Johnson - Corey's foray into carbocationic cascades - Interesting rearrangements - Overman and the Prins-Pinacol The Nonclassical Problem: Early Curiosities Wagner, 1899: 1,2 shift OH H - H+ Meerwein, 1922: 1,2 shift OH Meerwin, H. -
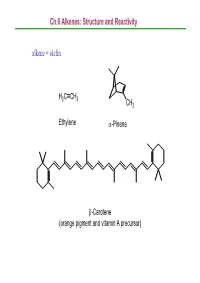
Ch.6 Alkenes: Structure and Reactivity Alkene = Olefin
Ch.6 Alkenes: Structure and Reactivity alkene = olefin H2CCH2 CH3 Ethylene α-Pinene β-Carotene (orange pigment and vitamin A precursor) Ch.6 Alkenes: Structure and Reactivity 6.1 Industrial Preparation and Use of Alkenes Compounds derived industrially from ethylene CH3CH2OH Ethanol CH3CHO Acetaldehyde CH3COOH Acetic acid HOCH2CH2OH Ethylene glycol ClCH2CH2Cl Ethylene dichloride H C=CHCl Vinyl chloride H2CCH2 2 O Ethylene oxide Ethylene (26 million tons / yr) O Vinyl acetate O Polyethylene Ch.6 Alkenes: Structure and Reactivity Compounds derived industrially from propylene OH Isopropyl alcohol H3CCH3 O Propylene oxide CH3 H3CCH CH2 Propylene Cumene (14 million tons / yr) CH3 CH3 Polypropylene Ch.6 Alkenes: Structure and Reactivity • Ethylene, propylene, and butene are synthesized industrially by thermal cracking of natural gas (C1-C4 alkanes) and straight-run gasoline (C4-C8 alkanes). 850-900oC CH (CH ) CH H + CH + H C=CH + CH CH=CH 3 2 n 3 steam 2 4 2 2 3 2 + CH3CH2CH=CH2 - the exact processes are complex; involve radical process H 900oC CH3CH2 CH2CH3 22H2CCH H2C=CH2 +H2 Ch.6 Alkenes: Structure and Reactivity • Thermal cracking is an example of a reaction whose energetics are dominated by entropy (∆So) rather than enthalpy (∆Ho) in the free-energy equation (∆Go = ∆Ho -T∆So) . ; C-C bond cleavage (positive ∆Ho) ; high T and increased number of molecules → larger T∆So Ch.6 Alkenes: Structure and Reactivity 6.2 Calculating Degree of Unsaturation unsaturated: formula of alkene CnH2n ; formula of alkane CnH2n+2 in general, each ring or double