George A. Olah 151
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DFT-Based Reaction Pathway Analysis of Hexadiene Cyclization Via Carbenium Ion Intermediates: Mechanistic Study of Light Alkane Aromatization Catalysis
J. Phys. Chem. B 2004, 108, 971-980 971 DFT-Based Reaction Pathway Analysis of Hexadiene Cyclization via Carbenium Ion Intermediates: Mechanistic Study of Light Alkane Aromatization Catalysis Yogesh V. Joshi, Aditya Bhan, and Kendall T. Thomson* School of Chemical Engineering, Purdue UniVersity, West Lafayette, Indiana 47907 ReceiVed: July 28, 2003; In Final Form: NoVember 4, 2003 We conducted density functional theory calculations to identify the complete cyclization mechanism (ring formation and ring expansion) for protonated hexadiene in the gas-phase as a precursory means of studying aromatization of light alkanes in acidic zeolite catalysts. We identify the rate-determining step to consist of ring expansion from a methylcyclopenta carbenium precursor to a stable cyclohexa carbenium intermediate, exhibiting an activation barrier of 9.6 kcal/mol and proceeding through a bicyclic intermediate starting from a secondary cyclopentyl carbocation. This pathway for ring closure was preferred over tertiary precursor expansion and direct cyclization to a cyclohexyl carbocation. Expecting carbocation intermediates to be represented by alkoxide species near Brønsted acid sites, we calculated the relative stability of primary, secondary, and tertiary alkoxide analogues to cyclopentyl carbocation intermediates involved in ring expansion and notice a reversal of stability relative to gas-phase carbocation stabilities (i.e., primary > secondary > tertiary stability). However, on the basis of the notion that transition state stability depends heavily -

Is There an Acid Strong Enough to Dissolve Glass? – Superacids
ARTICLE Is there an acid strong enough to dissolve glass? – Superacids For anybody who watched cartoons growing up, the word unit is based on how acids behave in water, however as acid probably springs to mind images of gaping holes being very strong acids react extremely violently in water this burnt into the floor by a spill, and liquid that would dissolve scale cannot be used for the pure ‘common’ acids (nitric, anything you drop into it. The reality of the acids you hydrochloric and sulphuric) or anything stronger than them. encounter in schools, and most undergrad university Instead, a different unit, the Hammett acidity function (H0), courses is somewhat underwhelming – sure they will react is often preferred when discussing superacids. with chemicals, but, if handled safely, where’s the drama? A superacid can be defined as any compound with an People don’t realise that these extraordinarily strong acids acidity greater than 100% pure sulphuric acid, which has a do exist, they’re just rarely seen outside of research labs Hammett acidity function (H0) of −12 [1]. Modern definitions due to their extreme potency. These acids are capable of define a superacid as a medium in which the chemical dissolving almost anything – wax, rocks, metals (even potential of protons is higher than it is in pure sulphuric acid platinum), and yes, even glass. [2]. Considering that pure sulphuric acid is highly corrosive, you can be certain that anything more acidic than that is What are Superacids? going to be powerful. What are superacids? Its all in the name – super acids are intensely strong acids. -

The Problem the Diols, Such As Ethane-1,2-Diol, Which Are The
The problem The diols, such as ethane-1,2-diol, which are the products of the reaction with cold dilute potassium manganate(VII), are themselves quite easily oxidised by manganate(VII) ions. That means that the reaction won't stop at this point unless the potassium manganate(VII) solution is very dilute, very cold, and preferably not under acidic conditions. If you are using hot concentrated acidified potassium manganate(VII) solution, what you finally end up with depends on the arrangement of groups around the carbon-carbon double bond. Writing a structural formula to represent any alkene The formula below represents a general alkene. In organic chemistry, the symbol R is used to represent hydrocarbon groups or hydrogen in a formula when you don't want to talk about specific compounds. If you use the symbol more than once in a formula (as here), the various groups are written as R1, R2, etc. In this particular case, the double bond is surrounded by four such groups, and these can be any combination of same or different - so they could be 2 hydrogens, a methyl and an ethyl, or 1 hydrogen and 3 methyls, or 1 hydrogen and 1 methyl and 1 ethyl and 1 propyl, or any other combination you can think of. In other words, this formula represents every possible simple alkene: The first stage of the extended oxidation The acidified potassium manganate(VII) solution oxidises the alkene by breaking the carbon-carbon double bond and replacing it with two carbon-oxygen double bonds. 1 The products are known as carbonyl compounds because they contain the carbonyl group, C=O. -

The Strongest Acid Christopher A
Chemistry in New Zealand October 2011 The Strongest Acid Christopher A. Reed Department of Chemistry, University of California, Riverside, California 92521, USA Article (e-mail: [email protected]) About the Author Chris Reed was born a kiwi to English parents in Auckland in 1947. He attended Dilworth School from 1956 to 1964 where his interest in chemistry was un- doubtedly stimulated by being entrusted with a key to the high school chemical stockroom. Nighttime experiments with white phosphorus led to the Headmaster administering six of the best. He obtained his BSc (1967), MSc (1st Class Hons., 1968) and PhD (1971) from The University of Auckland, doing thesis research on iridium organotransition metal chemistry with Professor Warren R. Roper FRS. This was followed by two years of postdoctoral study at Stanford Univer- sity with Professor James P. Collman working on picket fence porphyrin models for haemoglobin. In 1973 he joined the faculty of the University of Southern California, becoming Professor in 1979. After 25 years at USC, he moved to his present position of Distinguished Professor of Chemistry at UC-Riverside to build the Centre for s and p Block Chemistry. His present research interests focus on weakly coordinating anions, weakly coordinated ligands, acids, si- lylium ion chemistry, cationic catalysis and reactive cations across the periodic table. His earlier work included extensive studies in metalloporphyrin chemistry, models for dioxygen-binding copper proteins, spin-spin coupling phenomena including paramagnetic metal to ligand radical coupling, a Magnetochemi- cal alternative to the Spectrochemical Series, fullerene redox chemistry, fullerene-porphyrin supramolecular chemistry and metal-organic framework solids (MOFs). -

Course Material 2.Pdf
Reactive Intermediates Source: https://www.askiitians.com/iit-jee-chemistry/organic-chemistry/iupac- and-goc/reaction-intermediates/ Table of Content • Carbocations • Carbanions • Free Radicals • Carbenes • Arenium Ions • Benzynes Synthetic intermediate are stable products which are prepared, isolated and purified and subsequently used as starting materials in a synthetic sequence. Reactive intermediate, on the other hand, are short lived and their importance lies in the assignment of reaction mechanisms on the pathway from the starting substrate to stable products. These reactive intermediates are not isolated, but are detected by spectroscopic methods, or trapped chemically or their presence is confirmed by indirect evidence. • Carbocations Carbocations are the key intermediates in several reactions and particularly in nucleophilic substitution reactions. Structure of Carbocations : Generally, in the carbocations the positively charged carbon atom is bonded to three other atoms and has no nonbonding electrons. It is sp 2 hybridized with a planar structure and bond angles of about 120°. There is a + vacant unhybridized p orbital which in the case of CH 3 lies perpendicular to the plane of C—H bonds. Stability of Carbocations: There is an increase in carbocation stability with additional alkyl substitution. Thus one finds that addition of HX to three typical olefins decreases in the order (CH 3)2C=CH 2>CH 3—CH = CH 2 > CH 2 = CH 2. This is due to the relative stabilities of the carbocations formed in the rate determining step which in turn follows from the fact that the stability is increased by the electron releasing methyl group (+I), three such groups being more effective than two, and two more effective than one. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Superacid Chemistry
SUPERACID CHEMISTRY SECOND EDITION George A. Olah G. K. Surya Prakash Arpad Molnar Jean Sommer SUPERACID CHEMISTRY SUPERACID CHEMISTRY SECOND EDITION George A. Olah G. K. Surya Prakash Arpad Molnar Jean Sommer Copyright # 2009 by John Wiley & Sons, Inc. All rights reserved Published by John Wiley & Sons, Inc., Hoboken, New Jersey Published simultaneously in Canada No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. Requests to the Publisher for permission should be addressed to the Permissions Department, John Wiley & Sons, Inc., 111 River Street, Hoboken, NJ 07030, (201) 748-6011, fax (201) 748-6008, or online at http://www.wiley.com/go/permission. Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created or extended by sales representatives or written sales materials. The advice and strategies contained herein may not be suitable for your situation. -

Lecture Outline a Closer Look at Carbocation Stabilities and SN1/E1 Reaction Rates We Learned That an Important Factor in the SN
Lecture outline A closer look at carbocation stabilities and SN1/E1 reaction rates We learned that an important factor in the SN1/E1 reaction pathway is the stability of the carbocation intermediate. The more stable the carbocation, the faster the reaction. But how do we know anything about the stabilities of carbocations? After all, they are very unstable, high-energy species that can't be observed directly under ordinary reaction conditions. In protic solvents, carbocations typically survive only for picoseconds (1 ps - 10–12s). Much of what we know (or assume we know) about carbocation stabilities comes from measurements of the rates of reactions that form them, like the ionization of an alkyl halide in a polar solvent. The key to connecting reaction rates (kinetics) with the stability of high-energy intermediates (thermodynamics) is the Hammond postulate. The Hammond postulate says that if two consecutive structures on a reaction coordinate are similar in energy, they are also similar in structure. For example, in the SN1/E1 reactions, we know that the carbocation intermediate is much higher in energy than the alkyl halide reactant, so the transition state for the ionization step must lie closer in energy to the carbocation than the alkyl halide. The Hammond postulate says that the transition state should therefore resemble the carbocation in structure. So factors that stabilize the carbocation also stabilize the transition state leading to it. Here are two examples of rxn coordinate diagrams for the ionization step that are reasonable and unreasonable according to the Hammond postulate. We use the Hammond postulate frequently in making assumptions about the nature of transition states and how reaction rates should be influenced by structural features in the reactants or products. -

Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and Its Interactions with Supramolecular Systems
View metadata,Downloaded citation and from similar orbit.dtu.dk papers on:at core.ac.uk May 03, 2019 brought to you by CORE provided by Online Research Database In Technology Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Reynisson, Johannes Publication date: 2000 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Reynisson, J. (2000). Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems. Roskilde: Risø National Laboratory. Denmark. Forskningscenter Risoe. Risoe-R, No. 1191(EN) General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Jóhannes Reynisson OO O Risø National Laboratory, Roskilde, Denmark June 2000 1 Abstract Trioxatriangulenium (TOTA+, 4,8,12-trioxa-4,8,12,12c-tetrahydro- dibenzo[cd,mn]-pyrenylium) is a closed shell carbocation which is stable in its crystalline form and in polar solvents at ambient temperatures. -

The Legacy of George Olah Chemical Engineering
PP Periodica Polytechnica The Legacy of George Olah Chemical Engineering 61(4), pp. 301-307, 2017 https://doi.org/10.3311/PPch.11352 Creative Commons Attribution b Miklós Simonyi1* research article Received 27 June 2017; accepted after revision 11 August 2017 Abstract 1 Introduction The life of George Olah exemplifies the fate of Hungarian The New York Times wrote on March 14, 2017: „George scientific excellence in the 20th century. He was talented and A. Olah, a Hungarian-born scientist who won the Nobel Prize a hard worker, but he could not remain in his country and had in Chemistry in 1994 for his study of the chemical reactions of to seek a new life in the West. His life-long scientific interests carbon compounds, died on Wednesday at his home in Beverly developed in the Budapest Technical University helped him Hills, Calif. He was 89.” to overcome obstacles and he became a leading chemist of Once again, a man of outstanding excellence with world- worldwide fame. His qualities and flexibility in both scientific wide fame died abroad. Olah’s studies and career started in research and practical applications serve as a brilliant example Budapest, as vividly described in his autobiography [1]. He for generations to come. obtained a diploma in chemical engineering at the Budapest Technical University (BTU) in 1949 and joined the Organic Keywords Chemistry Institute founded by Professor Géza Zemplén in early achievements, accommodating to foreign norms, scientific 1913, as the first University Department for Organic Chemistry breakthrough, the super-acids, in the service of mankind in Hungary. -

Nonclassical Carbocations: from Controversy to Convention
Nonclassical Carbocations H H C From Controversy to Convention H H H A Stoltz Group Literature Meeting brought to you by Chris Gilmore June 26, 2006 8 PM 147 Noyes Outline 1. Introduction 2. The Nonclassical Carbocation Controversy - Winstein, Brown, and the Great Debate - George Olah and ending the discussion - Important nonclassical carbocations 3. The Nature of the Nonclassical Carbocation - The 3-center, 2-electron bond - Cleaving C-C and C-H σ−bonds - Intermediate or Transition state? Changing the way we think about carbocations 4. Carbocations, nonclassical intermediates, and synthetic chemistry - Biosynthetic Pathways - Steroids, by W.S. Johnson - Corey's foray into carbocationic cascades - Interesting rearrangements - Overman and the Prins-Pinacol Carbocations: An Introduction Traditional carbocation is a low-valent, trisubstituted electron-deficient carbon center: R superacid R R LG R R R R R R "carbenium LGH ion" - 6 valence e- - planar structure - empty p orbital Modes of stabilization: Heteroatomic Assistance π-bond Resonance σ-bond Participation R X H2C X Allylic Lone Pair Anchimeric Homoconjugation Hyperconjugation Non-Classical Resonance Assistance Stabilization Interaction Outline 1. Introduction 2. The Nonclassical Carbocation Controversy - Winstein, Brown, and the Great Debate - George Olah and ending the discussion - Important nonclassical carbocations 3. The Nature of the Nonclassical Carbocation - The 3-center, 2-electron bond - Cleaving C-C and C-H σ−bonds - Intermediate or Transition state? Changing the way we think about carbocations 4. Carbocations, nonclassical intermediates, and synthetic chemistry - Biosynthetic Pathways - Steroids, by W.S. Johnson - Corey's foray into carbocationic cascades - Interesting rearrangements - Overman and the Prins-Pinacol The Nonclassical Problem: Early Curiosities Wagner, 1899: 1,2 shift OH H - H+ Meerwein, 1922: 1,2 shift OH Meerwin, H. -
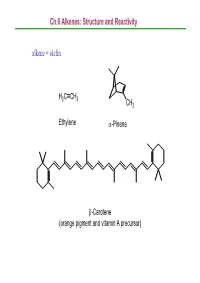
Ch.6 Alkenes: Structure and Reactivity Alkene = Olefin
Ch.6 Alkenes: Structure and Reactivity alkene = olefin H2CCH2 CH3 Ethylene α-Pinene β-Carotene (orange pigment and vitamin A precursor) Ch.6 Alkenes: Structure and Reactivity 6.1 Industrial Preparation and Use of Alkenes Compounds derived industrially from ethylene CH3CH2OH Ethanol CH3CHO Acetaldehyde CH3COOH Acetic acid HOCH2CH2OH Ethylene glycol ClCH2CH2Cl Ethylene dichloride H C=CHCl Vinyl chloride H2CCH2 2 O Ethylene oxide Ethylene (26 million tons / yr) O Vinyl acetate O Polyethylene Ch.6 Alkenes: Structure and Reactivity Compounds derived industrially from propylene OH Isopropyl alcohol H3CCH3 O Propylene oxide CH3 H3CCH CH2 Propylene Cumene (14 million tons / yr) CH3 CH3 Polypropylene Ch.6 Alkenes: Structure and Reactivity • Ethylene, propylene, and butene are synthesized industrially by thermal cracking of natural gas (C1-C4 alkanes) and straight-run gasoline (C4-C8 alkanes). 850-900oC CH (CH ) CH H + CH + H C=CH + CH CH=CH 3 2 n 3 steam 2 4 2 2 3 2 + CH3CH2CH=CH2 - the exact processes are complex; involve radical process H 900oC CH3CH2 CH2CH3 22H2CCH H2C=CH2 +H2 Ch.6 Alkenes: Structure and Reactivity • Thermal cracking is an example of a reaction whose energetics are dominated by entropy (∆So) rather than enthalpy (∆Ho) in the free-energy equation (∆Go = ∆Ho -T∆So) . ; C-C bond cleavage (positive ∆Ho) ; high T and increased number of molecules → larger T∆So Ch.6 Alkenes: Structure and Reactivity 6.2 Calculating Degree of Unsaturation unsaturated: formula of alkene CnH2n ; formula of alkane CnH2n+2 in general, each ring or double