Gut Microbiome Alterations in Ulcerative Colitis and After Moxibustion Intervention
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metaproteogenomic Insights Beyond Bacterial Response to Naphthalene
ORIGINAL ARTICLE ISME Journal – Original article Metaproteogenomic insights beyond bacterial response to 5 naphthalene exposure and bio-stimulation María-Eugenia Guazzaroni, Florian-Alexander Herbst, Iván Lores, Javier Tamames, Ana Isabel Peláez, Nieves López-Cortés, María Alcaide, Mercedes V. del Pozo, José María Vieites, Martin von Bergen, José Luis R. Gallego, Rafael Bargiela, Arantxa López-López, Dietmar H. Pieper, Ramón Rosselló-Móra, Jesús Sánchez, Jana Seifert and Manuel Ferrer 10 Supporting Online Material includes Text (Supporting Materials and Methods) Tables S1 to S9 Figures S1 to S7 1 SUPPORTING TEXT Supporting Materials and Methods Soil characterisation Soil pH was measured in a suspension of soil and water (1:2.5) with a glass electrode, and 5 electrical conductivity was measured in the same extract (diluted 1:5). Primary soil characteristics were determined using standard techniques, such as dichromate oxidation (organic matter content), the Kjeldahl method (nitrogen content), the Olsen method (phosphorus content) and a Bernard calcimeter (carbonate content). The Bouyoucos Densimetry method was used to establish textural data. Exchangeable cations (Ca, Mg, K and 10 Na) extracted with 1 M NH 4Cl and exchangeable aluminium extracted with 1 M KCl were determined using atomic absorption/emission spectrophotometry with an AA200 PerkinElmer analyser. The effective cation exchange capacity (ECEC) was calculated as the sum of the values of the last two measurements (sum of the exchangeable cations and the exchangeable Al). Analyses were performed immediately after sampling. 15 Hydrocarbon analysis Extraction (5 g of sample N and Nbs) was performed with dichloromethane:acetone (1:1) using a Soxtherm extraction apparatus (Gerhardt GmbH & Co. -

Achromobacter Buckle Infection Diagnosed by a 16S Rdna Clone
Hotta et al. BMC Ophthalmology 2014, 14:142 http://www.biomedcentral.com/1471-2415/14/142 CASE REPORT Open Access Achromobacter buckle infection diagnosed by a 16S rDNA clone library analysis: a case report Fumika Hotta1†, Hiroshi Eguchi1*, Takeshi Naito1†, Yoshinori Mitamura1†, Kohei Kusujima2† and Tomomi Kuwahara3† Abstract Background: In clinical settings, bacterial infections are usually diagnosed by isolation of colonies after laboratory cultivation followed by species identification with biochemical tests. However, biochemical tests result in misidentification due to similar phenotypes of closely related species. In such cases, 16S rDNA sequence analysis is useful. Herein, we report the first case of an Achromobacter-associated buckle infection that was diagnosed by 16S rDNA sequence analysis. This report highlights the significance of Achromobacter spp. in device-related ophthalmic infections. Case presentation: A 56-year-old woman, who had received buckling surgery using a silicone solid tire for retinal detachment eighteen years prior to this study, presented purulent eye discharge and conjunctival hyperemia in her right eye. Buckle infection was suspected and the buckle material was removed. Isolates from cultures of preoperative discharge and from deposits on the operatively removed buckle material were initially identified as Alcaligenes and Corynebacterium species. However, sequence analysis of a 16S rDNA clone library using the DNA extracted from the deposits on the buckle material demonstrated that all of the 16S rDNA sequences most closely matched those of Achromobacter spp. We concluded that the initial misdiagnosis of this case as an Alcaligenes buckle infection was due to the unreliability of the biochemical test in discriminating Achromobacter and Alcaligenes species due to their close taxonomic positions and similar phenotypes. -

Achromobacter Infections and Treatment Options
AAC Accepted Manuscript Posted Online 17 August 2020 Antimicrob. Agents Chemother. doi:10.1128/AAC.01025-20 Copyright © 2020 American Society for Microbiology. All Rights Reserved. 1 Achromobacter Infections and Treatment Options 2 Burcu Isler 1 2,3 3 Timothy J. Kidd Downloaded from 4 Adam G. Stewart 1,4 5 Patrick Harris 1,2 6 1,4 David L. Paterson http://aac.asm.org/ 7 1. University of Queensland, Faculty of Medicine, UQ Center for Clinical Research, 8 Brisbane, Australia 9 2. Central Microbiology, Pathology Queensland, Royal Brisbane and Women’s Hospital, 10 Brisbane, Australia on August 18, 2020 at University of Queensland 11 3. University of Queensland, Faculty of Science, School of Chemistry and Molecular 12 Biosciences, Brisbane, Australia 13 4. Infectious Diseases Unit, Royal Brisbane and Women’s Hospital, Brisbane, Australia 14 15 Editorial correspondence can be sent to: 16 Professor David Paterson 17 Director 18 UQ Center for Clinical Research 19 Faculty of Medicine 20 The University of Queensland 1 21 Level 8, Building 71/918, UQCCR, RBWH Campus 22 Herston QLD 4029 AUSTRALIA 23 T: +61 7 3346 5500 Downloaded from 24 F: +61 7 3346 5509 25 E: [email protected] 26 http://aac.asm.org/ 27 28 29 on August 18, 2020 at University of Queensland 30 31 32 33 34 35 36 37 38 39 2 40 Abstract 41 Achromobacter is a genus of non-fermenting Gram negative bacteria under order 42 Burkholderiales. Although primarily isolated from respiratory tract of people with cystic Downloaded from 43 fibrosis, Achromobacter spp. can cause a broad range of infections in hosts with other 44 underlying conditions. -

Identification, Molecular Epidemiology, and Antibiotic Resistance Characterization of Acinetobacter Spp
FACULTY OF HEALTH SCIENCES DEPARTMENT OF MEDICAL BIOLOGY UNIVERSITY HOSPITAL OF NORTH NORWAY DEPARTMENT OF MICROBIOLOGY AND INFECTION CONTROL REFERENCE CENTRE FOR DETECTION OF ANTIMICROBIAL RESISTANCE Identification, molecular epidemiology, and antibiotic resistance characterization of Acinetobacter spp. clinical isolates Nabil Karah A dissertation for the degree of Philosophiae Doctor June 2011 Acknowledgments The work presented in this thesis has been carried out between January 2009 and September 2011 at the Reference Centre for Detection of Antimicrobial Resistance (K-res), Department of Microbiology and Infection Control, University Hospital of North Norway (UNN); and the Research Group for Host–Microbe Interactions, Department of Medical Biology, Faculty of Health Sciences, University of Tromsø (UIT), Tromsø, Norway. I would like to express my deep and truthful acknowledgment to my main supervisor Ørjan Samuelsen. His understanding and encouraging supervision played a major role in the success of every experiment of my PhD project. Dear Ørjan, I am certainly very thankful for your indispensible contribution in all the four manuscripts. I am also very grateful to your comments, suggestions, and corrections on the present thesis. I am sincerely grateful to my co-supervisor Arnfinn Sundsfjord for his important contribution not only in my MSc study and my PhD study but also in my entire career as a “Medical Microbiologist”. I would also thank you Arnfinn for your nonstop support during my stay in Tromsø at a personal level. My sincere thanks are due to co-supervisors Kristin Hegstad and Gunnar Skov Simonsen for the valuable advice, productive comments, and friendly support. I would like to thank co-authors Christian G. -

A Comparison of Genospecies of Clinical Isolates in the Acinetobacter Spp. Complex Obtained from Hospitalized Patients in Busan, Korea
Biomedical Science Letters 2019, 25(1): 40 ~53 Original Article https://doi.org/10.15616/BSL.2019.25.1.40 eISSN : 2288-7415 A Comparison of Genospecies of Clinical Isolates in the Acinetobacter spp. Complex Obtained from Hospitalized Patients in Busan, Korea Gyu-Nam Park 1,*, Hye-Sook Kang 2,** , Hye-Ran Kim 3,** *, Bo-Kyung Jung 1,*, Do-Hee Kim 4,* and Kyung-Soo Chang 1, †,*** 1Department of Clinical Laboratory Science, College of Health Sciences, Catholic University of Pusan, Busan 46252, Korea 2Department of Laboratory Medicine, Maryknoll Medical Center, Busan 48972, Korea 3Department of Clinical Laboratory Science, College of Health and Therapy, Daegu Haany University, Gyeongsangbuk-Do 38610, Korea 4Department of Laboratory Medicine, Busan Veterans Hospital, Busan 46996, Korea Of the Acinetobacter spp., A. baumannii (genospecies 2) is the most clinically significant in terms of hospital-acquired infections worldwide. It is difficult to perform Acinetobacter -related taxonomy using phenotypic characteristics and routine laboratory methods owing to clusters of closely related species. The ability to accurately identify Acinetobacter spp. is clinically important because antimicrobial susceptibility and clinical relevance differs significantly among the different genospecies. Based on the medical importance of pathogenic Acinetobacter spp., the distribution and characterization of Acinetobacter spp. isolates from 123 clinical samples was determined in the current study using four typically applied bacterial identification methods; partial rpoB gene sequencing, amplified rRNA gene restriction analysis (ARDRA) of the intergenic transcribed spacer (ITS) region of the 16 ~23S rRNA, the VITEK ® 2 system (an automated microbial identification system) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). -

Detection of Acinetobacter Spp. in Blood Cultures by an Improved Fluorescent in Situ Hybridization Assay
Polish Journal of Microbiology 2018, Vol. 67, No 1, 3–10 ORIGINAL PAPER Detection of Acinetobacter spp. in Blood Cultures by an Improved Fluorescent in Situ Hybridization Assay HANIEH ASAADI1, 2, BEHROUZ NAEIMI1, 3, SOMAYYEH GHARIBI4, ABDALNASER KHOSRAVI1, SINA DOBARADARAN5, REZA TAHERKHANI1, 3 and SAEED TAJBAKHSH1, 3* 1 Department of Microbiology and Parasitology, Faculty of Medicine, Bushehr University of Medical Sciences, Bushehr, Iran 2 Student Research Committee, Bushehr University of Medical Sciences, Bushehr, Iran 3 The Persian Gulf Tropical Medicine Research Center, Bushehr University of Medical Sciences, Bushehr, Iran 4 Department of Microbiology, Faculty of Biological Sciences, Alzahra University, Tehran, Iran 5 Department of Environmental Health Engineering, Faculty of Health, Bushehr University of Medical Sciences, Bushehr, Iran Submitted 25 May 2017, revised 1 October 2017, accepted 28 November 2017 Abstract Fluorescent in situ hybridization (FISH) allows rapid detection of microorganisms. We aimed (i) to evaluate the sensitivity and specific- ity of FISH for the detection of Acinetobacter spp. in blood culture specimens and (ii) to test the simultaneous application of two genus- specific probes labeled with the same fluorochrome to increase the fluorescent signal intensity and improve the detection of Acinetobacter spp. Three hundred and twenty blood culture specimens were testedvia both the conventional laboratory methods and FISH to detect Acinetobacter spp. The specimens were examined separately with each genus-specific probe Aci and ACA, and also using a mixture of the both probes Aci and ACA. In all examinations, probe EUB338 was used accompanied by Aci and ACA. The specificity of FISH was 100% (97.5% confidence interval [CI] = 98.7% – 100%). -

The Genetic Analysis of an Acinetobacter Johnsonii Clinical Strain Evidenced the Presence of Horizontal Genetic Transfer
RESEARCH ARTICLE The Genetic Analysis of an Acinetobacter johnsonii Clinical Strain Evidenced the Presence of Horizontal Genetic Transfer Sabrina Montaña1, Sareda T. J. Schramm2, German Matías Traglia1, Kevin Chiem1,2, Gisela Parmeciano Di Noto1, Marisa Almuzara3, Claudia Barberis3, Carlos Vay3, Cecilia Quiroga1, Marcelo E. Tolmasky2, Andrés Iriarte4, María Soledad Ramírez1,2* 1 Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM, UBA-CONICET), Buenos Aires, Argentina, 2 Department of Biological Science, California State University Fullerton, Fullerton, CA, a11111 United States of America, 3 Laboratorio de Bacteriología Clínica, Departamento de Bioquímica Clínica, Hospital de Clínicas José de San Martín, Facultad de Farmacia y Bioquímica, Buenos Aires, Argentina, 4 Departamento de Desarrollo Biotecnológico, Instituto de Higiene, Facultad de Medicina, UdelaR, Montevideo, Uruguay * [email protected] OPEN ACCESS Abstract Citation: Montaña S, Schramm STJ, Traglia GM, Chiem K, Parmeciano Di Noto G, Almuzara M, et al. Acinetobacter johnsonii rarely causes human infections. While most A. johnsonii isolates are (2016) The Genetic Analysis of an Acinetobacter β johnsonii Clinical Strain Evidenced the Presence of susceptible to virtually all antibiotics, strains harboring a variety of -lactamases have Horizontal Genetic Transfer. PLoS ONE 11(8): recently been described. An A. johnsonii Aj2199 clinical strain recovered from a hospital in e0161528. doi:10.1371/journal.pone.0161528 Buenos Aires produces PER-2 and OXA-58. We decided to delve into its genome by obtain- Editor: Ruth Hall, University of Sydney, AUSTRALIA ing the whole genome sequence of the Aj2199 strain. Genome comparison studies on Received: March 23, 2016 Aj2199 revealed 240 unique genes and a close relation to strain WJ10621, isolated from the urine of a patient in China. -

Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales
Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales TECHNICAL BIOSAFETY COMMITTEE (TBC) NATIONAL CENTER FOR GENETIC ENGINEERING AND BIOTECHNOLOGY (BIOTEC) NATIONAL SCIENCE AND TECHNOLOGY DEVELOPMENT AGENCY (NSTDA) MINISTRY OF SCIENCE AND TECHNOLOGY (MOST) 2015 Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales TECHNICAL BIOSAFETY COMMITTEE (TBC) NATIONAL CENTER FOR GENETIC ENGINEERING AND BIOTECHNOLOGY (BIOTEC) NATIONAL SCIENCE AND TECHNOLOGY DEVELOPMENT AGENCY (NSTDA) MINISTRY OF SCIENCE AND TECHNOLOGY (MOST) 2015 Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales Technical Biosafety Committee National Center for Genetic Engineering and Biotechnology National Science and Technology Development Agency (NSTDA) © National Center for Genetic Engineering and Biotechnology 2015 ISBN : 978-616-12-0386-3 Tel : +66(0)2-564-6700 Fax : +66(0)2-564-6703 E-mail : [email protected] URL : http://www.biotec.or.th Printing House : P.A. Living Printing Co.,Ltd 4 Soi Sirintron 7 Road Sirintron District Bangplad Province Bangkok 10700 Tel : +66(0)2-881 9890 Fax : +66(0)2-881 9894 Preface Genetically Modified Microorganisms (GMMs) were first used in B.E. 2525 to produce insulin in industrial medicine. Currently, GMMs are used in various industries, such as the food, pharmaceutical and bioplastic industries, to manufacture a number of important consumer products. To ensure operator and environmental safety, the Technical Biosafety Committee (TBC) of the National Center for Genetic Engineering and Biotechnology (BIOTEC), the National Science and Technology Development Agency (NSTDA), has prepared guidelines for GMM work, publishing “Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales” in B.E. -

Contamination of Burn Wounds by Achromobacter
Annals of Burns and Fire Disasters - vol. XXIX - n. 3 - September 2016 CONTAMINATION OF BURN WOUNDS BY ACHROMOBACTER XYLOSOXIDANS FOLLOWED BY SEVERE INFECTION: 10-YEAR ANALYSIS OF A BURN UNIT POPULATION CONTAMINATION DES ZONES BRÛLÉES PAR ACHROMOBACTER XYLOSOXIDANS, ENTRAÎNANT UNE INFECTION SÉVÈRE: ANALYSE SUR 10 ANS * Schulz A., Perbix W., Fuchs P.C., Seyhan H., Schiefer J.L. Department of Plastic Surgery, Hand Surgery, Burn Center, University of Witten/Herdecke, Cologne-Merheim Medical Center (CMMC), Cologne, Germany SUMMARY. Gram-negative infections predominate in burn surgery. Until recently, Achromobacter species were described as sepsis-caus - ing bacteria in immunocompromised patients only. Severe infections associated with Achromobacter species in burn patients have been rarely reported. We retrospectively analyzed all burn patients in our database, who were treated at the Intensive Care Burn Unit (ICBU) of the Cologne Merheim Burn Centre from January 2006 to December 2015, focusing on contamination and infection by Achromobacter species. We identified 20 patients with burns contaminated by Achromobacter species within the 10-year study period. Four of these patients showed signs of infection concomitant with detection of Achromobacter species. Despite receiving complex antibiotic therapy based on antibiogram and resistogram typing, 3 of these patients, who had extensive burns, developed severe sepsis. Two patients ultimately died of multiple organ failure. In 1 case, Achromobacter xylosoxidans was the only isolate detected from the swabs and blood samples taken during the last stage of sepsis. Achromobacter xylosoxidans contamination of wounds of severely burned immunocompromised patients can lead to systemic lethal infection. Close monitoring of burn wounds for contamination by Achromobacter xylosoxidans is essential, and appropriate therapy must be administered as soon as possible. -

Long-Term Phenotypic Evolution of Bacteria
LETTER doi:10.1038/nature13827 Long-term phenotypic evolution of bacteria Germa´n Plata1,2, Christopher S. Henry3 & Dennis Vitkup1,4 For many decades comparative analyses of protein sequences and is sometimes observed within the same species, a transition from high structures have been used to investigate fundamental principles of to low phenotypic similarity occurs primarily at the genus level. molecular evolution1,2. In contrast, relatively little is known about Analyses of phenotypic evolution, such as the morphological varia- the long-term evolution of species’ phenotypic and genetic properties. tion of beaks in Darwin’s finches3, provided the original impetus and This represents an important gap in our understanding of evolu- context for understanding natural selection. Because the evolutionary tion, as exactly these proprieties play key roles in natural selection importance and physiological role of specific phenotypic traits change and adaptation to diverse environments. Here we perform a com- over time, it is often difficult to connect genotype to phenotype to fit- parative analysis of bacterial growth and gene deletion phenotypes ness across long evolutionary distances, especially for metazoan organ- using hundreds of genome-scale metabolic models. Overall, bacte- isms. For microbial species, on the other hand, the ability to metabolize rial phenotypic evolution can be described by a two-stage process different nutrient sources, although clearly not the only important phe- with a rapid initial phenotypic diversification followed by a slow notype, always remains an essential determinant of their fitness and long-term exponential divergence. The observed average divergence lifestyle. Even though a large-scale comparative analysis of microbial trend, with approximately similar fractions of phenotypic properties phenotypes—such as growth on different nutrients or the impact of changing per unit time, continues for billions of years. -

Alcaligenes Xylosoxidans Infections in Children Five Cases in Different Sites
Research Article Alcaligenes Xylosoxidans Infections in Children Five Cases in Different Sites AUTHORS: Sanz Santaeufemia FJ See correspondence Ramos Amador JT 1 [email protected] Muley Alonso R 2 [email protected] Bodas Pinedo A 4 [email protected] Hinojosa Mena-Bernal J 3 [email protected] García Talavera ME 5 [email protected] Department of Pediatrics. Hospital Niño Jesús. Madrid. 1. Inmunodeficiency Unit. 2. Pediatric Nephrology. 3. Pediatric Neurosurgery. Department of Pediatrics. Hospital 12 Octubre. Madrid. 4. Department of Pediatrics. Hospital Clinico Madrid. 5. Family Physician. Centro Salud Felipe II, Móstoles. Received date: 2 October 2013, Accepted date: 27 February 2014 Academic Editor: Angelika Lehner Correspondence and reprint requests: Fco José Sanz Santaeufemia, MD Pediatría Hospital Niño Jesús Avenida de Menéndez Pelayo 65 1 28009 Madrid, Spain ( 34-91-5035900. Ext 410 E-mail: [email protected] ABSTRACT Alcaligenes xylosoxidans, formerly known as Achromobacter xylosoxidans is a non- fermenting gram-negative rod, that is increasingly been identified as a pathogen in the last decade. Nowadays the name commonly accepted for correct taxonomy is Achromobacter xylosoxidans 1. It has been isolated from several aqueous environmental sources, some of which have been associated with nosocomial outbreaks of infections 2. Infections caused by Alcaligenes xylosoxidans have been documented in patients with a variety of indwelling devices, but it has been shown as a causing disease bacteria in other cases without risk factors (previous surgery or catheter carrier). It could be also encountered in all kind of organs and body systems, so this microorganism is acquiring major importance in recent years. -
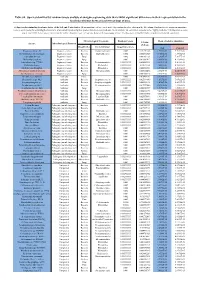
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp.