Transcription Elongation Rate Affects Nascent Histone Pre-Mrna Folding and 3′ End Processing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Network Assessment of Demethylation Treatment in Melanoma: Differential Transcriptome-Methylome and Antigen Profile Signatures
RESEARCH ARTICLE Network assessment of demethylation treatment in melanoma: Differential transcriptome-methylome and antigen profile signatures Zhijie Jiang1☯, Caterina Cinti2☯, Monia Taranta2, Elisabetta Mattioli3,4, Elisa Schena3,5, Sakshi Singh2, Rimpi Khurana1, Giovanna Lattanzi3,4, Nicholas F. Tsinoremas1,6, 1 Enrico CapobiancoID * a1111111111 1 Center for Computational Science, University of Miami, Miami, FL, United States of America, 2 Institute of Clinical Physiology, CNR, Siena, Italy, 3 CNR Institute of Molecular Genetics, Bologna, Italy, 4 IRCCS Rizzoli a1111111111 Orthopedic Institute, Bologna, Italy, 5 Endocrinology Unit, Department of Medical & Surgical Sciences, Alma a1111111111 Mater Studiorum University of Bologna, S Orsola-Malpighi Hospital, Bologna, Italy, 6 Department of a1111111111 Medicine, University of Miami, Miami, FL, United States of America a1111111111 ☯ These authors contributed equally to this work. * [email protected] OPEN ACCESS Abstract Citation: Jiang Z, Cinti C, Taranta M, Mattioli E, Schena E, Singh S, et al. (2018) Network assessment of demethylation treatment in Background melanoma: Differential transcriptome-methylome and antigen profile signatures. PLoS ONE 13(11): In melanoma, like in other cancers, both genetic alterations and epigenetic underlie the met- e0206686. https://doi.org/10.1371/journal. astatic process. These effects are usually measured by changes in both methylome and pone.0206686 transcriptome profiles, whose cross-correlation remains uncertain. We aimed to assess at Editor: Roger Chammas, Universidade de Sao systems scale the significance of epigenetic treatment in melanoma cells with different met- Paulo, BRAZIL astatic potential. Received: June 20, 2018 Accepted: October 17, 2018 Methods and findings Published: November 28, 2018 Treatment by DAC demethylation with 5-Aza-2'-deoxycytidine of two melanoma cell lines Copyright: © 2018 Jiang et al. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Genome-Wide Screen of Cell-Cycle Regulators in Normal and Tumor Cells
bioRxiv preprint doi: https://doi.org/10.1101/060350; this version posted June 23, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion Maria Sokolova1, Mikko Turunen1, Oliver Mortusewicz3, Teemu Kivioja1, Patrick Herr3, Anna Vähärautio1, Mikael Björklund1, Minna Taipale2, Thomas Helleday3 and Jussi Taipale1,2,* 1Genome-Scale Biology Program, P.O. Box 63, FI-00014 University of Helsinki, Finland. 2Science for Life laboratory, Department of Biosciences and Nutrition, Karolinska Institutet, SE- 141 83 Stockholm, Sweden. 3Science for Life laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, S-171 21 Stockholm, Sweden To identify cell cycle regulators that enable cancer cells to replicate DNA and divide in an unrestricted manner, we performed a parallel genome-wide RNAi screen in normal and cancer cell lines. In addition to many shared regulators, we found that tumor and normal cells are differentially sensitive to loss of the histone genes transcriptional regulator CASP8AP2. In cancer cells, loss of CASP8AP2 leads to a failure to synthesize sufficient amount of histones in the S-phase of the cell cycle, resulting in slowing of individual replication forks. Despite this, DNA replication fails to arrest, and tumor cells progress in an elongated S-phase that lasts several days, finally resulting in death of most of the affected cells. -

Multi-Sample Full-Length Transcriptome Analysis of 22 Breast Cancer Clinical
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Multi-sample Full-length Transcriptome Analysis of 22 Breast Cancer Clinical Specimens with Long-Read Sequencing Shinichi Namba1, §, Toshihide Ueno1, Shinya Kojima1, Yosuke Tanaka1, Satoshi Inoue1, Fumishi Kishigami1, Noriko Maeda2, Tomoko Ogawa3, Shoichi Hazama4, Yuichi Shiraishi5, Hiroyuki Mano1, and Masahito Kawazu1* Author affiliations: 1Division of Cellular Signaling, 5Division of Genome Analysis Platform Development, Research Institute, National Cancer Center, Tokyo 104-0045, Japan; 2Department of Gastroenterological, Breast and Endocrine Surgery, 4Department of Translational Research and Developmental Therapeutics against Cancer, Yamaguchi University Graduate School of Medicine, Yamaguchi 755-8505, Japan; 3Department of Breast Surgery, Mie University Hospital, Mie 514-8507, Japan; § Present address, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan; * Corresponding author E-mail:[email protected] (MK) bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Although transcriptome alteration is considered as one of the essential drivers of carcinogenesis, conventional short-read RNAseq technology has limited researchers from directly exploring full-length transcripts, only focusing on individual splice sites. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -
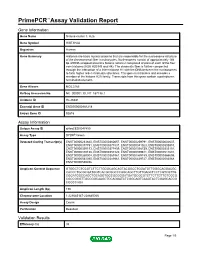
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 3, H2a Gene Symbol HIST3H2A Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A H2B H3 and H4). The chromatin fiber is further compacted through the interaction of a linker histone H1 with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene contain a palindromic termination element. Gene Aliases MGC3165 RefSeq Accession No. NC_000001.10, NT_167186.1 UniGene ID Hs.26331 Ensembl Gene ID ENSG00000181218 Entrez Gene ID 92815 Assay Information Unique Assay ID qHsaCED0047450 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233840, ENST00000360657, ENST00000259791, ENST00000602637, ENST00000377791, ENST00000377831, ENST00000341023, ENST00000303910, ENST00000359193, ENST00000377459, ENST00000358739, ENST00000333151, ENST00000330180, ENST00000357320, ENST00000359611, ENST00000511601, ENST00000260008, ENST00000369161, ENST00000369159, ENST00000366695, ENST00000543095, ENST00000389961, ENST00000439727, ENST00000583968, ENST00000580456 Amplicon Context Sequence GTGGCTCTCCGTCTTCTTGGGCAGCAGTACGGCCTGGATGTTGGGCAGGACGC CACCCTGCGCGATGGTCACGCGGCCCAGCAGCTTGTTGAGCTCCTCGTCGTTG CGGATGGCCAGCTGCAGGTGGCGCGGGATGATGCGCGTCTTCTTGTTGTCGCG -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder
INVESTIGATION A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder Aparna Prasad,* Daniele Merico,* Bhooma Thiruvahindrapuram,* John Wei,* Anath C. Lionel,*,† Daisuke Sato,* Jessica Rickaby,* Chao Lu,* Peter Szatmari,‡ Wendy Roberts,§ Bridget A. Fernandez,** Christian R. Marshall,*,†† Eli Hatchwell,‡‡ Peggy S. Eis,‡‡ and Stephen W. Scherer*,†,††,1 *The Centre for Applied Genomics, Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto M5G 1L7, Canada, †Department of Molecular Genetics, University of Toronto, Toronto M5G 1L7, Canada, ‡Offord Centre for Child Studies, Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton L8P 3B6, § Canada, Autism Research Unit, The Hospital for Sick Children, Toronto M5G 1X8, Canada, **Disciplines of Genetics and Medicine, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3V6, Canada, ††McLaughlin Centre, University of Toronto, Toronto M5G 1L7, Canada, and ‡‡Population Diagnostics, Inc., Melville, New York 11747 ABSTRACT The identification of rare inherited and de novo copy number variations (CNVs) in human KEYWORDS subjects has proven a productive approach to highlight risk genes for autism spectrum disorder (ASD). A rare variants variety of microarrays are available to detect CNVs, including single-nucleotide polymorphism (SNP) arrays gene copy and comparative genomic hybridization (CGH) arrays. Here, we examine a cohort of 696 unrelated ASD number cases using a high-resolution one-million feature CGH microarray, the majority of which were previously chromosomal genotyped with SNP arrays. Our objective was to discover new CNVs in ASD cases that were not detected abnormalities by SNP microarray analysis and to delineate novel ASD risk loci via combined analysis of CGH and SNP array cytogenetics data sets on the ASD cohort and CGH data on an additional 1000 control samples. -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 RESEARCH ARTICLE A Mutation in Histone H2B Represents a New Class of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,4, Rajarshi P. Ghosh5, Adrian T. Grzybowski6, Tao Yu7, Qiao Zhang8, Alberto Riva9, Tanmay P. Lele8, George C. Schatz4, Neil L. Kelleher4, Alexander J. Ruthenburg6, Jan Liphardt5, and Jonathan D. Licht1 Downloaded from cancerdiscovery.aacrjournals.org on September 30, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 ABSTRACT By examination of the cancer genomics database, we identified a new set of mutations in core histones that frequently recur in cancer patient samples and are predicted to disrupt nucleosome stability. In support of this idea, we characterized a glutamate to lysine mutation of histone H2B at amino acid 76 (H2B-E76K), found particularly in bladder and head and neck cancers, that disrupts the interaction between H2B and H4. Although H2B-E76K forms dimers with H2A, it does not form stable histone octamers with H3 and H4 in vitro, and when recon- stituted with DNA forms unstable nucleosomes with increased sensitivity to nuclease. Expression of the equivalent H2B mutant in yeast restricted growth at high temperature and led to defective nucleosome-mediated gene repression. Significantly, H2B-E76K expression in the normal mammary epithelial cell line MCF10A increased cellular proliferation, cooperated with mutant PIK3CA to pro- mote colony formation, and caused a significant drift in gene expression and fundamental changes in chromatin accessibility, particularly at gene regulatory elements. -

Loss of Neuronal 3D Chromatin Organization Causes Transcriptional and Behavioural Deficits Related to Serotonergic Dysfunction
ARTICLE Received 22 Jan 2014 | Accepted 18 Jun 2014 | Published 18 Jul 2014 DOI: 10.1038/ncomms5450 Loss of neuronal 3D chromatin organization causes transcriptional and behavioural deficits related to serotonergic dysfunction Satomi Ito1,*,w, Adriana Magalska2,*, Manuel Alcaraz-Iborra1, Jose P. Lopez-Atalaya1, Victor Rovira1, Bruno Contreras-Moreira3, Michal Lipinski1, Roman Olivares1, Jose Martinez-Hernandez4, Blazej Ruszczycki2, Rafael Lujan4, Emilio Geijo-Barrientos1, Grzegorz M. Wilczynski2 & Angel Barco1 The interior of the neuronal cell nucleus is a highly organized three-dimensional (3D) structure where regions of the genome that are linearly millions of bases apart establish sub-structures with specialized functions. To investigate neuronal chromatin organization and dynamics in vivo, we generated bitransgenic mice expressing GFP-tagged histone H2B in principal neurons of the forebrain. Surprisingly, the expression of this chimeric histone in mature neurons caused chromocenter declustering and disrupted the association of heterochromatin with the nuclear lamina. The loss of these structures did not affect neuronal viability but was associated with specific transcriptional and behavioural deficits related to serotonergic dysfunction. Overall, our results demonstrate that the 3D organization of chromatin within neuronal cells provides an additional level of epigenetic regulation of gene expression that critically impacts neuronal function. This in turn suggests that some loci associated with neuropsychiatric disorders may be particularly sensitive to changes in chromatin architecture. 1 Instituto de Neurociencias (Universidad Miguel Herna´ndez—Consejo Superior de Investigaciones Cientı´ficas), Avenida Santiago Ramo´n y Cajal s/n, Sant Joan d’Alacant, 03550 Alicante, Spain. 2 Nencki Institute of Experimental Biology, Pasteura 3, 02-093 Warsaw, Poland. -

Identification of Genetic Factors Underpinning Phenotypic Heterogeneity in Huntington’S Disease and Other Neurodegenerative Disorders
Identification of genetic factors underpinning phenotypic heterogeneity in Huntington’s disease and other neurodegenerative disorders. By Dr Davina J Hensman Moss A thesis submitted to University College London for the degree of Doctor of Philosophy Department of Neurodegenerative Disease Institute of Neurology University College London (UCL) 2020 1 I, Davina Hensman Moss confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Collaborative work is also indicated in this thesis. Signature: Date: 2 Abstract Neurodegenerative diseases including Huntington’s disease (HD), the spinocerebellar ataxias and C9orf72 associated Amyotrophic Lateral Sclerosis / Frontotemporal dementia (ALS/FTD) do not present and progress in the same way in all patients. Instead there is phenotypic variability in age at onset, progression and symptoms. Understanding this variability is not only clinically valuable, but identification of the genetic factors underpinning this variability has the potential to highlight genes and pathways which may be amenable to therapeutic manipulation, hence help find drugs for these devastating and currently incurable diseases. Identification of genetic modifiers of neurodegenerative diseases is the overarching aim of this thesis. To identify genetic variants which modify disease progression it is first necessary to have a detailed characterization of the disease and its trajectory over time. In this thesis clinical data from the TRACK-HD studies, for which I collected data as a clinical fellow, was used to study disease progression over time in HD, and give subjects a progression score for subsequent analysis. In this thesis I show blood transcriptomic signatures of HD status and stage which parallel HD brain and overlap with Alzheimer’s disease brain.