Fine-Mapping Quantitative Trait Loci Affecting Murine External Ear Tissue Regeneration in the LG/J by SM/J Advanced Intercross Line
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -
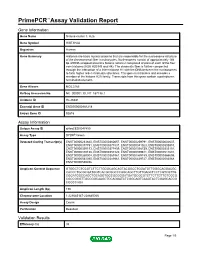
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 3, H2a Gene Symbol HIST3H2A Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A H2B H3 and H4). The chromatin fiber is further compacted through the interaction of a linker histone H1 with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene contain a palindromic termination element. Gene Aliases MGC3165 RefSeq Accession No. NC_000001.10, NT_167186.1 UniGene ID Hs.26331 Ensembl Gene ID ENSG00000181218 Entrez Gene ID 92815 Assay Information Unique Assay ID qHsaCED0047450 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233840, ENST00000360657, ENST00000259791, ENST00000602637, ENST00000377791, ENST00000377831, ENST00000341023, ENST00000303910, ENST00000359193, ENST00000377459, ENST00000358739, ENST00000333151, ENST00000330180, ENST00000357320, ENST00000359611, ENST00000511601, ENST00000260008, ENST00000369161, ENST00000369159, ENST00000366695, ENST00000543095, ENST00000389961, ENST00000439727, ENST00000583968, ENST00000580456 Amplicon Context Sequence GTGGCTCTCCGTCTTCTTGGGCAGCAGTACGGCCTGGATGTTGGGCAGGACGC CACCCTGCGCGATGGTCACGCGGCCCAGCAGCTTGTTGAGCTCCTCGTCGTTG CGGATGGCCAGCTGCAGGTGGCGCGGGATGATGCGCGTCTTCTTGTTGTCGCG -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder
INVESTIGATION A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder Aparna Prasad,* Daniele Merico,* Bhooma Thiruvahindrapuram,* John Wei,* Anath C. Lionel,*,† Daisuke Sato,* Jessica Rickaby,* Chao Lu,* Peter Szatmari,‡ Wendy Roberts,§ Bridget A. Fernandez,** Christian R. Marshall,*,†† Eli Hatchwell,‡‡ Peggy S. Eis,‡‡ and Stephen W. Scherer*,†,††,1 *The Centre for Applied Genomics, Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto M5G 1L7, Canada, †Department of Molecular Genetics, University of Toronto, Toronto M5G 1L7, Canada, ‡Offord Centre for Child Studies, Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton L8P 3B6, § Canada, Autism Research Unit, The Hospital for Sick Children, Toronto M5G 1X8, Canada, **Disciplines of Genetics and Medicine, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3V6, Canada, ††McLaughlin Centre, University of Toronto, Toronto M5G 1L7, Canada, and ‡‡Population Diagnostics, Inc., Melville, New York 11747 ABSTRACT The identification of rare inherited and de novo copy number variations (CNVs) in human KEYWORDS subjects has proven a productive approach to highlight risk genes for autism spectrum disorder (ASD). A rare variants variety of microarrays are available to detect CNVs, including single-nucleotide polymorphism (SNP) arrays gene copy and comparative genomic hybridization (CGH) arrays. Here, we examine a cohort of 696 unrelated ASD number cases using a high-resolution one-million feature CGH microarray, the majority of which were previously chromosomal genotyped with SNP arrays. Our objective was to discover new CNVs in ASD cases that were not detected abnormalities by SNP microarray analysis and to delineate novel ASD risk loci via combined analysis of CGH and SNP array cytogenetics data sets on the ASD cohort and CGH data on an additional 1000 control samples. -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 RESEARCH ARTICLE A Mutation in Histone H2B Represents a New Class of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,4, Rajarshi P. Ghosh5, Adrian T. Grzybowski6, Tao Yu7, Qiao Zhang8, Alberto Riva9, Tanmay P. Lele8, George C. Schatz4, Neil L. Kelleher4, Alexander J. Ruthenburg6, Jan Liphardt5, and Jonathan D. Licht1 Downloaded from cancerdiscovery.aacrjournals.org on September 30, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 ABSTRACT By examination of the cancer genomics database, we identified a new set of mutations in core histones that frequently recur in cancer patient samples and are predicted to disrupt nucleosome stability. In support of this idea, we characterized a glutamate to lysine mutation of histone H2B at amino acid 76 (H2B-E76K), found particularly in bladder and head and neck cancers, that disrupts the interaction between H2B and H4. Although H2B-E76K forms dimers with H2A, it does not form stable histone octamers with H3 and H4 in vitro, and when recon- stituted with DNA forms unstable nucleosomes with increased sensitivity to nuclease. Expression of the equivalent H2B mutant in yeast restricted growth at high temperature and led to defective nucleosome-mediated gene repression. Significantly, H2B-E76K expression in the normal mammary epithelial cell line MCF10A increased cellular proliferation, cooperated with mutant PIK3CA to pro- mote colony formation, and caused a significant drift in gene expression and fundamental changes in chromatin accessibility, particularly at gene regulatory elements. -

The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights Into Cell Differentiation and Function
International Journal of Molecular Sciences Article The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function Yashwanth Subbannayya 1 , Markus Haug 1, Sneha M. Pinto 1, Varshasnata Mohanty 2, Hany Zakaria Meås 1, Trude Helen Flo 1, T.S. Keshava Prasad 2 and Richard K. Kandasamy 1,* 1 Centre of Molecular Inflammation Research (CEMIR), Department of Clinical and Molecular Medicine (IKOM), Norwegian University of Science and Technology, 7491 Trondheim, Norway; [email protected] (Y.S.); [email protected] (M.H.); [email protected] (S.M.P.); [email protected] (H.Z.M.); trude.fl[email protected] (T.H.F.) 2 Center for Systems Biology and Molecular Medicine, Yenepoya (Deemed to be University), Mangalore 575018, India; [email protected] (V.M.); [email protected] (T.S.K.P.) * Correspondence: [email protected] Abstract: CD4+ T cells (T helper cells) are cytokine-producing adaptive immune cells that activate or regulate the responses of various immune cells. The activation and functional status of CD4+ T cells is important for adequate responses to pathogen infections but has also been associated with auto-immune disorders and survival in several cancers. In the current study, we carried out a label-free high-resolution FTMS-based proteomic profiling of resting and T cell receptor-activated (72 h) primary human CD4+ T cells from peripheral blood of healthy donors as well as SUP-T1 cells. We identified 5237 proteins, of which significant alterations in the levels of 1119 proteins were observed between resting and activated CD4+ T cells. -

Differential Expression Profile Prioritization of Positional Candidate Glaucoma Genes the GLC1C Locus
LABORATORY SCIENCES Differential Expression Profile Prioritization of Positional Candidate Glaucoma Genes The GLC1C Locus Frank W. Rozsa, PhD; Kathleen M. Scott, BS; Hemant Pawar, PhD; John R. Samples, MD; Mary K. Wirtz, PhD; Julia E. Richards, PhD Objectives: To develop and apply a model for priori- est because of moderate expression and changes in tization of candidate glaucoma genes. expression. Transcription factor ZBTB38 emerges as an interesting candidate gene because of the overall expres- Methods: This Affymetrix GeneChip (Affymetrix, Santa sion level, differential expression, and function. Clara, Calif) study of gene expression in primary cul- ture human trabecular meshwork cells uses a positional Conclusions: Only1geneintheGLC1C interval fits our differential expression profile model for prioritization of model for differential expression under multiple glau- candidate genes within the GLC1C genetic inclusion in- coma risk conditions. The use of multiple prioritization terval. models resulted in filtering 7 candidate genes of higher interest out of the 41 known genes in the region. Results: Sixteen genes were expressed under all condi- tions within the GLC1C interval. TMEM22 was the only Clinical Relevance: This study identified a small sub- gene within the interval with differential expression in set of genes that are most likely to harbor mutations that the same direction under both conditions tested. Two cause glaucoma linked to GLC1C. genes, ATP1B3 and COPB2, are of interest in the con- text of a protein-misfolding model for candidate selec- tion. SLC25A36, PCCB, and FNDC6 are of lesser inter- Arch Ophthalmol. 2007;125:117-127 IGH PREVALENCE AND PO- identification of additional GLC1C fami- tential for severe out- lies7,18-20 who provide optimal samples for come combine to make screening candidate genes for muta- adult-onset primary tions.7,18,20 The existence of 2 distinct open-angle glaucoma GLC1C haplotypes suggests that muta- (POAG) a significant public health prob- tions will not be limited to rare descen- H1 lem. -

Noncoding Rnas As Novel Pancreatic Cancer Targets
NONCODING RNAS AS NOVEL PANCREATIC CANCER TARGETS by Amy Makler A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, FL August 2018 Copyright 2018 by Amy Makler ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous support, constant encouragement, and his gentle, but sometimes critical, guidance throughout the past two years of my master’s education. His faith in my abilities and his belief in my future success ensured I continue down this path of research. Working in Dr. Narayanan’s lab has truly been an unforgettable experience as well as a critical step in my future endeavors. I would also like to extend my gratitude to my committee members, Dr. Binninger and Dr. Jia, for their support and suggestions regarding my thesis. Their recommendations added a fresh perspective that enriched our initial hypothesis. They have been indispensable as members of my committee, and I thank them for their contributions. My parents have been integral to my successes in life and their support throughout my education has been crucial. They taught me to push through difficulties and encouraged me to pursue my interests. Thank you, mom and dad! I would like to thank my boyfriend, Joshua Disatham, for his assistance in ensuring my writing maintained a logical progression and flow as well as his unwavering support. He was my rock when the stress grew unbearable and his encouraging words kept me pushing along. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,†
bioRxiv preprint doi: https://doi.org/10.1101/720235; this version posted July 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,† Abstract Core histone proteins H2A, H2B, H3, and H4 are encoded by a large family of genes dis- tributed across the human genome. Canonical core histones contribute the majority of proteins to bulk chromatin packaging, and are encoded in 4 clusters by 65 coding genes comprising 17 for H2A, 18 for H2B, 15 for H3, and 15 for H4, along with at least 17 total pseudogenes. The canonical core histone genes display coding variation that gives rise to 11 H2A, 15 H2B, 4 H3, and 2 H4 unique protein isoforms. Although histone proteins are highly conserved overall, these isoforms represent a surprising and seldom recognised variation with amino acid identity as low as 77 % between canonical histone proteins of the same type. The gene sequence and protein isoform diversity also exceeds com- monly used subtype designations such as H2A.1 and H3.1, and exists in parallel with the well-known specialisation of variant histone proteins. RNA sequencing of histone transcripts shows evidence for differential expression of histone genes but the functional significance of this variation has not yet been investigated. To assist understanding of the implications of histone gene and protein diversity we have catalogued the entire human canonical core histone gene and protein complement. -

RNF7 (1-113, His-Tag) Human Protein – AR51110PU-S | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AR51110PU-S RNF7 (1-113, His-tag) Human Protein Product data: Product Type: Recombinant Proteins Description: RNF7 (1-113, His-tag) human recombinant protein, 0.1 mg Species: Human Expression Host: E. coli Tag: His-tag Predicted MW: 15.1 kDa Concentration: lot specific Purity: >90% by SDS - PAGE Buffer: Presentation State: Purified State: Liquid purified protein Buffer System: 20 mM Tris-HCl buffer (pH 8.0) containing 10% glycerol, 0.4M Urea Preparation: Liquid purified protein Protein Description: Recombinant human RNF7 protein, fused to His-tag at N-terminus, was expressed in E.coli . Storage: Store undiluted at 2-8°C for one week or (in aliquots) at -20°C to -80°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. RefSeq: NP_001188299 Locus ID: 9616 UniProt ID: Q9UBF6 Cytogenetics: 3q23 Synonyms: CKBBP1; rbx2; ROC2; SAG Summary: The protein encoded by this gene is a highly conserved ring finger protein. It is an essential subunit of SKP1-cullin/CDC53-F box protein ubiquitin ligases, which are a part of the protein degradation machinery important for cell cycle progression and signal transduction. This protein interacts with, and is a substrate of, casein kinase II (CSNK2A1/CKII). The phosphorylation of this protein by CSNK2A1 has been shown to promote the degradation of IkappaBalpha (CHUK/IKK-alpha/IKBKA) and p27Kip1(CDKN1B). Alternatively spliced transcript variants encoding distinct isoforms have been reported.