Structural Relaxation Around Substitutional Cr in Pyrope Garnet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

About Our Mineral World
About Our Mineral World Compiled from series of Articles titled "TRIVIAL PURSUITS" from News Nuggets by Paul F. Hlava "The study of the natural sciences ought to expand the mind and enlarge the ability to grasp intellectual problems." Source?? "Mineral collecting can lead the interested and inquisitive person into the broader fields of geology and chemistry. This progression should be the proper outcome. Collecting for its own sake adds nothing to a person's understanding of the world about him. Learning to recognize minerals is only a beginning. The real satisfaction in mineralogy is in gaining knowledge of the ways in which minerals are formed in the earth, of the chemistry of the minerals and of the ways atoms are packed together to form crystals. Only by grouping minerals into definite categories is is possible to study, describe, and discuss them in a systematic and intelligent manner." Rock and Minerals, 1869, p. 260. Table of Contents: AGATE, JASPER, CHERT AND .............................................................................................................................2 GARNETS..................................................................................................................................................................2 GOLD.........................................................................................................................................................................3 "The Mystery of the Magnetic Dinosaur Bones" .......................................................................................................4 -

Nomenclature of the Garnet Supergroup
American Mineralogist, Volume 98, pages 785–811, 2013 IMA REPORT Nomenclature of the garnet supergroup EDWARD S. GREW,1,* ANDREW J. LOCOCK,2 STUART J. MILLS,3,† IRINA O. GALUSKINA,4 EVGENY V. GALUSKIN,4 AND ULF HÅLENIUS5 1School of Earth and Climate Sciences, University of Maine, Orono, Maine 04469, U.S.A. 2Department of Earth and Atmospheric Sciences, University of Alberta, Edmonton, Alberta T6G 2E3, Canada 3Geosciences, Museum Victoria, GPO Box 666, Melbourne 3001, Victoria, Australia 4Faculty of Earth Sciences, Department of Geochemistry, Mineralogy and Petrography, University of Silesia, Będzińska 60, 41-200 Sosnowiec, Poland 5Swedish Museum of Natural History, Department of Mineralogy, P.O. Box 50 007, 104 05 Stockholm, Sweden ABSTRACT The garnet supergroup includes all minerals isostructural with garnet regardless of what elements occupy the four atomic sites, i.e., the supergroup includes several chemical classes. There are pres- ently 32 approved species, with an additional 5 possible species needing further study to be approved. The general formula for the garnet supergroup minerals is {X3}[Y2](Z3)ϕ12, where X, Y, and Z refer to dodecahedral, octahedral, and tetrahedral sites, respectively, and ϕ is O, OH, or F. Most garnets are cubic, space group Ia3d (no. 230), but two OH-bearing species (henritermierite and holtstamite) have tetragonal symmetry, space group, I41/acd (no. 142), and their X, Z, and ϕ sites are split into more symmetrically unique atomic positions. Total charge at the Z site and symmetry are criteria for distinguishing groups, whereas the dominant-constituent and dominant-valency rules are critical in identifying species. Twenty-nine species belong to one of five groups: the tetragonal henritermierite group and the isometric bitikleite, schorlomite, garnet, and berzeliite groups with a total charge at Z of 8 (silicate), 9 (oxide), 10 (silicate), 12 (silicate), and 15 (vanadate, arsenate), respectively. -

Garnets from the Camafuca-Camazambo Kimberlite (Angola)
Anais da Academia Brasileira de Ciências (2006) 78(2): 309-315 (Annals of the Brazilian Academy of Sciences) ISSN 0001-3765 www.scielo.br/aabc Garnets from the Camafuca-Camazambo kimberlite (Angola) EUGÉNIO A. CORREIA and FERNANDO A.T.P. LAIGINHAS Departamento de Geologia, Faculdade de Ciências da Universidade do Porto Praça de Gomes Teixeira, 4050 Porto, Portugal Manuscript received on November 17, 2004; accepted for publication on June 13, 2005; presented by ALCIDES N. SIAL ABSTRACT This work presents a geochemical study of a set of garnets, selected by their colors, from the Camafuca- Camazambo kimberlite, located on northeast Angola. Mantle-derived garnets were classified according to the scheme proposed by Grütter et al. (2004) and belong to the G1, G4, G9 and G10 groups. Both sub-calcic (G10) and Ca-saturated (G9) garnets, typical, respectively, of harzburgites and lherzolites, were identified. The solubility limit of knorringite molecule in G10D garnets suggests they have crystallized at a minimum pressure of about 40 to 45 kbar (4– 4.5 GPa). The occurrence of diamond stability field garnets (G10D) is a clear indicator of the potential of this kimberlite for diamond. The chemistry of the garnets suggests that the source for the kimberlite was a lherzolite that has suffered a partial melting that formed basaltic magma, leaving a harzburgite as a residue. Key words: kimberlite, diamond, garnet, lherzolite, harzburgite. INTRODUCTION GEOCHEMICAL STUDY OF GARNETS The Camafuca-Camazambo kimberlite belongs to The geochemical study of mantle-derived garnets a kimberlite province comprising over a dozen of occurring in kimberlites progressed remarkably in primary occurrences of diamond, located along- recent years, as their chemistry is not only a possible side the homonymous brook which is a tributary of indicator of the presence of diamonds in their host the Chicapa river, in the proximity of the Calonda rock but also their abundance is a likely indicator of village, northeast Angola (Fig. -

Partitioning of Chromium Between Garnet and Clinopyroxene: first-Principle Modelling Versus Metamorphic Assemblages
Eur. J. Mineral., 32, 387–403, 2020 https://doi.org/10.5194/ejm-32-387-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Partitioning of chromium between garnet and clinopyroxene: first-principle modelling versus metamorphic assemblages Sarah Figowy, Benoît Dubacq, Yves Noël, and Philippe d’Arco Sorbonne Université, CNRS-INSU, Institut des Sciences de la Terre de Paris, ISTeP UMR 7193, 75005 Paris, France Correspondence: Sarah Figowy (sarah.fi[email protected]) Received: 30 December 2019 – Revised: 28 May 2020 – Accepted: 15 June 2020 – Published: 3 July 2020 Abstract. Understanding the geochemical behaviour of trace and minor elements in mineral assemblages is of primary importance to study small- and large-scale geological processes. Partition coefficients are frequently used to model the chemical evolution of minerals and fluids during melting and in metamorphic rocks of all grades. However, kinetic effects hampering equilibrium partitioning may invalidate the modelling. This study aims at calculating partition coefficients and testing their applicability in natural mineral assemblages, choosing Cr in garnet and clinopyroxene via exchange with Al as a case study. First-principle modelling has been com- bined with measurements and element mapping to estimate partition coefficients for Cr and the deviation from equilibrium. Results highlight the role of crystal chemistry over the strain field around point defects, controlling the dynamics of the Cr3C D Al3C exchange between clinopyroxene and garnet. Ab initio calculations allowed estimation of Cr partition coefficients between garnet and clinopyroxene, using a thermodynamic approach based on endmembers and mixing models simplified for trace element behaviour. -

Thermodynamic Modelling of Cr-Bearing Garnets with Implications for Diamond Inclusions and Peridotite Xenoliths
Lithos 112S (2009) 986–991 Contents lists available at ScienceDirect Lithos journal homepage: www.elsevier.com/locate/lithos Thermodynamic modelling of Cr-bearing garnets with implications for diamond inclusions and peridotite xenoliths S. Klemme a,⁎, T.J. Ivanic b,e, J.A.D. Connolly d, B. Harte b,c a Institut für Mineralogie, Corrensstr. 24, Universität Münster, 48149 Münster, Germany b School of GeoSciences, King's Buildings, West Mains Road, University of Edinburgh, Edinburgh EH9 3JW, United Kingdom c Centre for Science at Extreme Conditions, University of Edinburgh, Erskine Williamson Building, Mayfield Road, Edinburgh EH9 3JZ, United Kingdom d Institut für Mineralogie und Petrographie, ETH Zentrum, Sonneggstrasse 5, CH-8082, Zürich, Switzerland e Geological Survey of Western Australia, Mineral House, 100 Plain Street, East Perth, WA 6004, Australia article info abstract Article history: A new approach is presented for modelling Cr-rich peridotite compositions and garnet–spinel compositions Received 8 September 2008 found in diamonds and xenoliths at conditions relevant to the deep continental Earth. Using recent Accepted 10 May 2009 experimental data, it is now possible to calculate phase relations and mineral compositions relevant to Available online 6 June 2009 pressures, temperatures, and compositions of the deep lithospheric Earth using free energy minimization techniques. Here we present calculated phase relations in Cr-rich mantle compositions from pressures of 20– Keywords: 60 kbar, and temperatures 800–1400 °C. The model is successful at modelling a wide range of natural mineral Garnet–spinel transition Peridotite compositions which are found as xenoliths in diamond-bearing kimberlites from South Africa, and is Diamond inclusion illustrated using suites of Cr-rich xenoliths from near Kimberley, South Africa. -

A New Chromium Garnet End Member, Knorringite, from Kimberlite
THB AMERICAN MINDRALOGIST, VOL.53, NOVEMBER DECEMBER, 1968 A NEW CHROMIUM GARNET END MEMBER, KNORRINGITE, FROM KIMBERLITE PnrBn H. NrxoNl aNl GeoncB HonnuNc, Departmentof Earth Sciences,Uniaersity of Leed.s,Leed,s, England. Assrnect Analysis by electron probe confirms that a garnet from Kao kimberlite pipe in Lesotho contains a large proportion of the component Mg3Cr2(SiO4)s. This is now called knorringite and thus replaces the name hanleite which has been discredited (:uvarovite). The garnet from Kao, with 17.4TaCrsos, is green, o: 11.65,n:1.803, G (obs):3.756, G (calc) 3.852. The properties of this and other "chromium pyropes" from kimberlites are described and their significance discussed. INrnoouctrorq Pyrope garnetscontaining chromium have long been known to occur in kimberlites (Wagner, 1914; Williams, 1932). They characterisethe ultrabasic nodules of these bodies, chrome-spinel peridotites, their higher level equivalents being much less common. (Nixon et al., 1963). Typically pyropes contain about 2-3 percent CrgOaequivalent to 5 to 8 percent of the uvarovite molecule,but greater amounts are found. A partial analysis of chromium pyrope from E9 lherzolite nodule from Liqhobong, Lesotho, gave 5.14 percent CrzOr and a full analysis of chromium pyrope from E10 serpentinisednodule from Sekameng,Leso- tho gave 7.52 percent Cr2Os(table 1) (Nixon et aI., 1963). E10 garnet contains8.7 percent MgsCrr(SiO+)a as calculatedin terms of end members of the garnets, there being insufficient calcium to match the high chrome content. If one calculates all the chromium as magnesium chromium garnet instead of calculatingfirst the uvarovite molecule,then consider- ably more of the new componentis seento be present. -

Knorringite-Uvarovite Garnet and Cr-Eskola Pyroxene in Ureilite Lew 88774
64th Annual Meteoritical Society Meeting (2001) 5016.pdf KNORRINGITE-UVAROVITE GARNET AND CR-ESKOLA PYROXENE IN UREILITE LEW 88774. C.A. Goodrich1 and G.E. Harlow2. 1Max-Planck-Institut für Chemie, PO 3060, D-55020 Mainz, Germany ([email protected]). 2American Museum of Natural History, New York, NY 10024 USA. Introduction: In LEW 88774 chromite grains (~100-800 µm) are enclosed by oikocrysts of orthopyroxene with exsolved augite [1-5]. They are generally associated with patches of graphite and commonly also with olivine. All grains have 5-40 µm-wide rims of glass (~70-75% SiO2, ~13-18% Al2O3), with corroded edges against the glass, and are surrounded by an assemblage of unusual Cr-rich minerals including Fe,Cr-carbide(s), Fe,Cr-sulfide(s), and eskolaite-corundum [1,2]. We describe two new members of this assemblage: knorringite-uvarovite garnet, and what appears to be a Cr-rich pyroxene that would be an Mg-rich Cr-analog of Ca-Eskola pyroxene. The latter should be a new mineral. Cr-rich Garnet and Pyroxene: These minerals form rims or bands (10-20 µm wide) at the contact between glass-rimmed chromite grains and the surrounding silicates. Where the surrounding silicate is olivine, the rims/bands consist of the Cr-rich garnet. Where the surrounding silicate is opx, they consist of the Cr-rich pyroxene. Where the surrounding silicate is augite, they are absent. The garnet contains 27% Cr2O3 and has the structural formula: (Mg3.7-3.8Fe0.2-0.4Ca1.7-1.8)5.9-6.1(Cr3.2-3.5Al0.4-0.7Ti0.1)3.8-4.0Si6O24. -

1 Refractive Indices of Minerals and Synthetic Compounds Ruth C. Shannon Geological Sciences
1 2 Refractive Indices of Minerals and Synthetic Compounds 3 4 Ruth C. Shannon 5 Geological Sciences/ CIRES, University of Colorado, Boulder, Colorado 80309 6 Barbara Lafuente 7 NASA Ames Research Center, Moffett Field, Mountain View, CA 94035 8 Robert D. Shannon 9 Geological Sciences/ CIRES, University of Colorado, Boulder, Colorado 80309 10 Robert T. Downs 11 Department of Geosciences, University of Arizona, 1040 E. 4th St, Tucson, Arizona 85721-0077 12 Reinhard X. Fischer 13 Universität Bremen, FB 5 Geowissenschaften, Klagenfurter Str. 2, D-28359 Bremen (Germany) 14 15 Abstract 16 This is a comprehensive compilation of refractive indices of 1933 minerals and 1019 synthetic 17 compounds including exact chemical compositions and references taken from 30 compilations 18 and many mineral and synthetic oxide descriptions. It represents a subset of about 4000 entries 19 used by Shannon and Fischer (Amer. Mineral. 101, 2016, 2288-2300) to determine the 20 polarizabilities of 270 cations and anions after removing 425 minerals and compounds 21 containing the lone-pair ions (Tl+, Sn2+, Pb2+, As3+, Sb3+,Bi3+, S4+, Se4+, Te4+, Cl5+, Br5+, I5+) and 22 uranyl ions, U6+. The table lists the empirical composition of the mineral or synthetic compound, 23 the ideal composition of the mineral, the mineral name or synthetic compound, the Dana classes 24 and subclasses extended to include beryllates, aluminates, gallates, germanates, niobates, 25 tantalates, molybdates, tungstates, etc., descriptive notes, e.g. structure polytypes and other 1 26 information -

RUBY in DIAMOND by Henry 0.A, Meyer and Edward Gubelin
RUBY IN DIAMOND By Henry 0.A, Meyer and Edward Gubelin The first substantiated identification of corundum It was also observed during the early micro- (var. ruby) occurring as an inclusion in natural probe analyses conducted by Meyer and Boyd diamond is presented. The ruby is assigned to the (1968, 1969, 19721, and described by Sobolev (1974) eclogitic suite of inclusions in diamond, and the and by Prinz et al. (1975), that not only did the implications of its occurrence are discussed in garnets comprise two distinct groups, but most relation to the genesis of "eclogitic" diar~londs.It is inclusions in diamond could be assigned to one concluded that diamond crystallizes from a melt over a long period of time with possible fluctuations of two suites as well: the ultramafic * or the eclo- in ambient temperature and geochemical gitic*. The members of these suites are listed in environment. table 1. Reviews of the minerals in these suites are to be found in Meyer and Tsai (1976))Gubelin et al. (19781, and Harris and Gurney (1979). It should be noted that ruby does not appear on Inclusions in gemstones can be fascinating for either list of inclusions in table 1. scientist and layman alike. Scientifically, they aid in deciphering the genesis of the mineral; gemo- RUBY INCLUSION IN DIAMOND logically, they are often distinctive and aid in During endeavors to find diamonds with unusual, identifying the host stone (Gubelin, 1953, 1974). colored mineral inclusions, one of the authors re- In 1645, John Evelyn, a diarist, reported seeing ceived two diamonds of unknown origin with red a "faire Rubie" inside a diamond that belonged to crystal inclusions that, on the basis of previous a Venetian nobleman (DeBeer, 1955). -

Crystal Structure of Synthetic Mg3cr2si3o12, the High-Pressure Cr End-Member of the Knorringite-Pyrope Garnet Series
Crystal structure of synthetic Mg3Cr2Si3O12, the high-pressure Cr end-member of the knorringite-pyrope garnet series Amélie Juhin 1, Guillaume Morin 1, Erik Elkaim 2, Dan J. Frost 3, Farid Juillot 1, Georges Calas 1 1 Institut de Minéralogie et de Physique des Milieux Condensés, UMR CNRS 7590 / Université Paris 6 / Université Paris 7 / IPGP - 140 rue de Lourmel 75015 Paris, France 2 Synchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin - BP 48 91192 Gif-sur-Yvette Cedex, France 3 Bayerisches Geoinstitut, Universität Bayreuth D-95440 Bayreuth, Germany ABSTRACT Knorringite, the Cr-end-member of the pyrope garnet series (Nixon et al. 1968), often occur in high proportions in kimberlite garnets and is thus used for tracing high-pressure deep-earth conditions favorable to the formation of diamonds, in which knorringite-rich garnet can occur as inclusions. However, although the synthesis of knorringite is reported in the literature (Ringwood 1977; Irifune et al. 1982; Taran et al. 2004), the structure of the pure end-member has not been yet determined from experimental data. In this study, the crystal structure of knorringite, Mg3Cr2(SiO4)3, has been refined from high resolution synchrotron X-ray powder diffraction data recorded under ambient conditions on a polycrystalline sample synthesized at 12 GPa in a multi-anvil apparatus. The structure is cubic, space 3 -3 group Ia-3d, a = 11.5935(1), V = 1558.27(4) Å , dcalc = 3.97 g.cm . The Cr-O distance of 1.957(2) Å is consistent with EXAFS results on the same sample. This short distance indicates a substantial 3+ compression of the CrO6 octahedron, compared to ambient pressure Cr -minerals such as uvarovite (<Cr- O> = 1.99 Å, Andrut and Wildner 2002). -
Petrography and Mineral Chemistry of Ultramafic and Related Inclusions from the Orapa A/K-1 Kimberlite Pipe, Botswana by Richard P
PETROGRAPHY AND MINERAL CHEMISTRY OF ULTRAMAFIC AND RELATED INCLUSIONS FROM THE ORAPA A/K-1 KIMBERLITE PIPE, BOTSWANA BY RICHARD P. TOLLO '-- Lesotho (/) ~ Q,) Q,) -E 0 0 I{) 500 meters generalized cross-section of a kimberlite pipe modified after Hawthorne ( 197 5) CONTRIBUTION NO. 39 DEPARTMENT OF GEOLOGY AND GEOGRAPHY UNIVERSITY OF MASSACHUSETIS AMHERST, MASSACHUSETIS PETROGRAPHY AND MINERAL CHEMISTRY OF ULTRAMAFIC AND RELATED INCLUSIONS FROM THE ORAPA A/K-1 KIMBERLITE PIPE, BOTSWANA by RICHARD PAUL TOLLO Contribution No. 39 Department of GeoJogy and Geography University of Massachusetts Amherst, Massachusetts August, 1982 paAJaSaE S4q~lE TTV OtTOl tnad p~aqOJE TABLE OF CONTENTS ACKNOWLEDGEMENT xi ABSTRACT . xiii Chapter I. INTRODUCTION 1 The Orapa A/K-1 Kimberlite Pipe 1 Determinative Methods 3 II. OXIDE DISCRETE NODULES .. 6 Ilmenite . • • • • • . • • • 6 Rutile and Ilmenite-Rutile Intergrowths 33 III. SILICATE DISCRETE NODULES 65 Clinopyroxene • • • • 65 Ilmenite-Clinopyroxene Intergrowth 79 Garnet . 86 Knorringite Garnet-Clinopyroxene-Chromite Intergrowth • • • • • . • • . • . • • . • . 101 IV. ECLOGITE NODULES • 112 Introduction • • 112 Petrography 112 Phase Chemistry 121 Geothermometry and Geobarometry 150 V. DISCUSSION •.. 158 VI. SUMMARY A}ID CONCLUSIONS 177 REFERENCES 182 APPENDIX A: LISTING OF THE INDIVIDUAL ELECTRON MICROPROBE ANALYSES USED IN THE CONSTRUCTION OF FIGURE 7 196 APPENDIX B: SUMMARY OF THE EXPERIMENTAL CONDITIONS INVOLVED IN THE ILMENITE-RUTILE SYNTHESIS EXPERIMENTS . • • . 202 iii LIST OF TABLES Table lo Average chemical compositions of individual ilmenite discrete nodules from Orapa - nodule interiors 18 2o Chemical compositions of coexisting ilmenite and rutile in ilmenite-rutile intergrowth nodules from Orapa and Jagersfontein o o o o o 0 o o o o o • • o • • 0 42 3. -
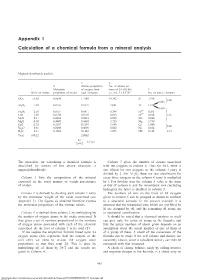
Appendix 1 Calculation of a Chemical Formula from a Mineral Analysis
Appendix 1 Calculation of a chemical formula from a mineral analysis Appendix 1 Magnesiohornblende analysis 3 4 2 Atomic proportion No. of anions on 1 Molecular of oxygen from basis of 24 (O,OH) 5 Wt.% of oxides proportion of oxides each molecule i.e. col. 368.3735 No. of ions in formula SiO 51.63 0.8594 1.7188 14.392 Si 7.196 2 8.00 0.804 } Al2O3 7.39 0.0725 0.2175 1.821 Al 1.214 0.410 3+ Fe2O3 2.50 0.0157 0.0471 0.394 Fe 0.263 FeO 5.30 0.0738 0.0738 0.618 Fe2+ 0.618 5.07 MnO 0.17 0.0024 0.0024 0.020 Mn 0.020 } MgO 18.09 0.4489 0.4489 3.759 Mg 3.759 CaO 12.32 0.2197 0.2197 1.840 Ca 1.840 2.00 Na2O 0.61 0.0098 0.0098 0.082 Na 0.164 } H2O+ 2.31 0.1282 0.1282 1.073 OH 2.146 2.15 Total 100.32 2.8662 24 = 8.3735 2.8662 The procedure for calculating a chemical formula is Column 5 gives the number of cations associated described by means of the above example, a with the oxygens in column 4. Thus for SiO2 there is magnesiohornblende. one silicon for two oxygens so the column 4 entry is divided by 2. For A12O3 there are two aluminiums for Column 1 lists the composition of the mineral every three oxygens so the column 4 entry is multiplied expressed in the usual manner as weight percentages by ~˜.