An Experimental and Theoretical Study of Reaction Steps Relevant to the Methanol-To-Hydrocarbons Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Disproportionation and Transalkylation of Alkylbenzenes Over Zeolite Catalysts
Applied Catalysis A: General 181 (1999) 355±398 Disproportionation and transalkylation of alkylbenzenes over zeolite catalysts Tseng-Chang Tsaia, Shang-Bin Liub, Ikai Wangc,* aRe®ning and Manufacturing Research Center, Chinese Petroleum Corporation, Chiayi 600, Taiwan bInstitute of Atomic and Molecular Sciences, Academia Sinica, PO Box 23-166, Taipei 106, Taiwan cDepartment of Chemical Engineering, National Tsing-Hua University, Hsinchu 300, Taiwan Received 13 June 1998; received in revised form 3 October 1998; accepted 5 November 1998 Abstract Disproportionation and transalkylation are important processes for the interconversion of mono-, di-, and tri-alkylbenzenes. In this review, we discuss the recent advances in process technology with special focus on improvements of para-isomer selectivity and catalyst stability. Extensive patent search and discussion on technology development are presented. The key criteria for process development are identi®ed. The working principles of para-isomer selectivity improvements involve the reduction of diffusivity and the inactivation of external surface. In conjunction with the fundamental research, various practical modi®cation aspects particularly the pre-coking and the silica deposition techniques, are extensively reviewed. The impact of para-isomer selective technology on process economics and product recovery strategy is discussed. Furthermore, perspective trends in related research and development are provided. # 1999 Elsevier Science B.V. All rights reserved. Keywords: Disproportionation; Transalkylation; -

Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules
Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Cite as: Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 Published Online: 15 October 2009 Sharon G. Lias, Joel F. Liebman, and Rhoda D. Levin ARTICLES YOU MAY BE INTERESTED IN Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update Journal of Physical and Chemical Reference Data 27, 413 (1998); https:// doi.org/10.1063/1.556018 Density-functional thermochemistry. III. The role of exact exchange The Journal of Chemical Physics 98, 5648 (1993); https://doi.org/10.1063/1.464913 A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu The Journal of Chemical Physics 132, 154104 (2010); https://doi.org/10.1063/1.3382344 Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 13, 695 © 1984 American Institute of Physics for the National Institute of Standards and Technology. Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Sharon G. Lias Center for Chemical Physics, National Bureau of Standards, Gaithersburg, MD 20899 Joel F. Liebman Department of Chemistry, University of Maryland Baltimore County, Catonsville, MD 21228 and Rhoda D. Levin Center for Chemical Physics. National Bureau of Standards, Gaithersburg, MD 20899 The available data on gas phase basicities and proton affinities of molecules are compiled and evaluated. Tables giving the molecules ordered (1) according to proton affinity and (2) according to empirical formula, sorted alphabetically are provided. -

Alkyne Metathesis Catalysts: Scope and Future André Mortreux, Olivier Coutelier
Alkyne Metathesis Catalysts: Scope And Future André Mortreux, Olivier Coutelier To cite this version: André Mortreux, Olivier Coutelier. Alkyne Metathesis Catalysts: Scope And Future. Journal of Molecular Catalysis A: Chemical, Elsevier, 2006, 254, pp.96-104. 10.1016/j.molcata.2006.03.054. hal-00107451 HAL Id: hal-00107451 https://hal.archives-ouvertes.fr/hal-00107451 Submitted on 18 Oct 2006 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ALKYNE METATHESIS CATALYSTS: SCOPE AND FUTURE. André Mortreux*, Olivier Coutelier Laboratoire de Catalyse de Lille UMR 8010 CNRS USTL-ENSCL, BP 90108, 59652 Villeneuve d’Ascq Cedex France Corresponding author.Tel +33320434993 ;Fax+33320434486 E-mail adress: [email protected] ABSTRACT This paper presents the evolution of alkyne metathesis since the early discoveries, essentially from the catalyst point of view. It is shown that although well defined carbynes may be useful for this reaction, further work has been made, aimed at the synthesis of new catalysts or catalytic systems, based on molybdenum precursors , associated or not with phenolic co- catalysts. The major objectives have been to obtain more functional groups tolerants catalysts, for their application in organic synthesis, including RCM for further stereoselective hydrogenation of the triple bond in the cycle, as well as for polymerization of aromatic diynes. -

Gas Phase Alkylation-Liquid Phase Transalkylation Process
~™ IIINMNNIIMIIIINIIINNII (19) J European Patent Office Office europeen des brevets (11) EP 0 879 809 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) |nt. CI.6: C07C 15/073, C07C 2/66 25.11.1998 Bulletin 1998/48 //B01J29/035 (21) Application number: 98109045.9 (22) Date of filing: 19.05.1998 (84) Designated Contracting States: • Merrill, James T. AT BE CH CY DE DK ES Fl FR GB GR IE IT LI LU Katy, Texas 77449 (US) MCNLPTSE • Butler, James R. Designated Extension States: Houston, Texas 77059 (US) AL LT LV MK RO SI (74) Representative: (30) Priority: 21.05.1997 US 861206 Leyder, Francis et al c/o Fina Research S.A. (71) Applicant: FINA TECHNOLOGY, INC. Dept. Brevets Dallas, Texas 75206 (US) Zone Industrielle C 7181 Seneffe (Feluy) (BE) (72) Inventors: • Ghosh, Ashim Kumar Houston, Texas 77059 (US) (54) Gas phase alkylation-liquid phase transalkylation process (57) Process for the production of ethylbenzene by aromatic fraction is subject to disproportionation to pro- alkylation over a silicalite alkylation catalyst with the vide a reduced diethylbenzene content and an subsequent transalkylation of diethylbenzene with the enhanced ethylbenzene content. A specific monoclinic alkylation catalyst and conditions selected to retard silicalite alkylation catalyst has a silica/alumina ratio of xylene production and also heavies production. A feed- at least 300 and has a crystal size of less than one stock containing benzene and ethylene is applied to a micron. multi-stage alkylation reaction zone having a plurality of series-connected catalyst beds containing a pentasil molecular sieve alkylation catalyst which is silicalite of a predominantly monoclinic symmetry having a silica/alu- mina ratio of at least 275. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -
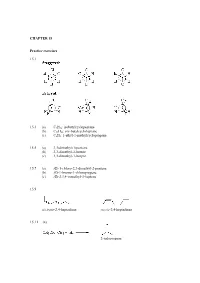
CHAPTER 15 Practice Exercises 15.1 15.3 (A) C9H18
CHAPTER 15 Practice exercises 15.1 15.3 (a) C9H18: isobutylcyclopentane (b) C11H22: sec-butylcycloheptane (c) C6H2: 1-ethyl-1-methylcyclopropane 15.5 (a) 3,3-dimethyl-1-pentene (b) 2,3-dimethyl-2-butene (c) 3,3-dimethyl-1-butyne 15.7 (a) (E)-1-chloro-2,3-dimethyl-2-pentene (b) (Z)-1-bromo-1-chloropropene (c) (E)-2,3,4-trimethyl-3-heptene 15.9 cis,trans-2,4-heptadiene cis,cis-2,4-heptadiene 15.11 (a) 2-iodopropane (b) 1-iodo-1-methylcyclohexane 15.13 CH3 CH3 + HI I Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step + CH3 HI CH3 I rate-determining step Step 2: Nucleophilic attack of the iodide anion on the 3˚ carbocation intermediate to give the product: CH + 3 CH3 I I 15.15 H2O CH3 CH3 + H3O OH Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step H + + CH3 HOH CH3 O H rate-determining H step Step 2: Nucleophilic attack of the water on the 3˚ carbocation intermediate to give the protonated alcohol: H H O+ H + CH3 O H CH3 Step 3: The protonated alcohol loses a proton to form the product: H + O H H CH3 + O + H3O CH3 H OH 15.17 (a) 2-phenyl-2-propanol (b) (E)-3,4-diphenyl-3-hexene (c) 3-methylbenzoic acid or m-methylbenzoic acid Review questions 15.1 A hydrocarbon is a compound composed only of hydrogen and carbon atoms. 15.3 In saturated hydrocarbons, each carbon is bonded to four other atoms, either hydrogen or carbon atoms. -

Monitoring the Structure–Reactivity Relationship in Epoxidized Perilla and Safflower Oil Thermosetting Resins
Monitoring the structure–reactivity relationship in epoxidized perilla and safflower oil thermosetting resins Thi-Nguyet Tran, Chiara Di Mauro, Alain Graillot, Alice Mija To cite this version: Thi-Nguyet Tran, Chiara Di Mauro, Alain Graillot, Alice Mija. Monitoring the structure–reactivity relationship in epoxidized perilla and safflower oil thermosetting resins. Polymer Chemistry, Royal Society of Chemistry - RSC, 2020, 11 (31), pp.5088-5097. 10.1039/D0PY00688B. hal-02902528v2 HAL Id: hal-02902528 https://hal.archives-ouvertes.fr/hal-02902528v2 Submitted on 27 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Polymer Chemistry PAPER Monitoring the structure–reactivity relationship in epoxidized perilla and safflower oil thermosetting Cite this: Polym. Chem., 2020, 11, 5088 resins† Thi-Nguyet Tran,a Chiara Di Mauro,a Alain Graillot b and Alice Mija *a For the first time, the effect of reactant structure, stoichiometry and heating rate on the reactivity of epox- idized perilla oil (EPLO) and epoxidized safflower oil (ESFO) with dicarboxylic acids (DCAs) was studied using in situ FT-IR. The epoxy content in the monomer structure was found to affect the copolymeriza- tion system’s reactivity, with epoxidized linseed oil (ELO) considered as a reference. -

Rearrangement Reactions
Rearrangement Reactions A rearrangement reaction is a broad class of organic reactions where the carbon skeleton of a molecule is rearranged to give a structural isomer of the original molecule. 1, 2-Rearrangements A 1, 2-rearrangement is an organic reaction where a substituent moves from one atom to another atom in a chemical compound. In a 1, 2 shift the movement involves two adjacent atoms but moves over larger distances are possible. In general straight-chain alkanes, are converted to branched isomers by heating in the presence of a catalyst. Examples include isomerisation of n-butane to isobutane and pentane to isopentane. Highly branched alkanes have favorable combustion characteristics for internal combustion engines. Further examples are the Wagner-Meerwein rearrangement: and the Beckmann rearrangement, which is relevant to the production of certain nylons: Pericyclic reactions A pericyclic reaction is a type of reaction with multiple carbon-carbon bonds making and breaking wherein the transition state of the molecule has a cyclic geometry and the reaction progresses in a concerted fashion. Examples are hydride shifts [email protected] and the Claisen rearrangement: Olefin metathesis Olefin metathesis is a formal exchange of the alkylidene fragments in two alkenes. It is a catalytic reaction with carbene, or more accurately, transition metal carbene complexintermediates. In this example (ethenolysis, a pair of vinyl compounds form a new symmetrical alkene with expulsion of ethylene. Pinacol rearrangement The pinacol–pinacolone rearrangement is a method for converting a 1,2-diol to a carbonyl compound in organic chemistry. The 1,2-rearrangement takes place under acidic conditions. -

1 the Principle of Conservation of Structural Aspect: Facilitating Visual Communication and Learning in Organic Chemistry Instr
1 The Principle of Conservation of Structural Aspect: Facilitating Visual Communication and Learning in Organic Chemistry Instruction John Andraos CareerChem 504-1129 Don Mills Road Toronto, ON M3B 2W4 Canada [email protected] Abstract An effective pedagogical method is presented for the visual communication of chemical reactions learned in organic chemistry undergraduate courses. The basis for the method is the preservation of the visual aspect of reactant and product structures so that the tracking of cleaved and formed chemical bonds is made self-evident. This consequently leads to improved clarity of presentation and a better understanding and grasp of proposed reaction mechanisms to explain product outcomes. The method is demonstrated for a variety of individual reaction types and synthesis plans. Various visual training exercises are also presented using ChemDraw Ultra 7.0 software and literature table of contents (TOC) graphics appearing in journal articles. Keywords: upper-division undergraduate, curriculum, organic chemistry pedagogy, problem solving, organic synthesis Introduction It is well known that when students encounter organic chemistry for the first time in undergraduate courses, they perceive the subject as “hard”. From their point of view their complaint stems from two major issues: (a) the apparent overwhelming volume of material that needs to be digested and learned in a relatively short period of time 2 (typically a three-month course session); and (b) the material is presented in a visual manner that is not always “easy” or evident to understand what is going in any given chemical reaction that is presented to them. The inevitable outcome is increased frustration and ultimately a dislike for the subject for the majority of students, particularly those that take organic chemistry as a standard mandatory requirement for other disciplines such as pre-medical studies and life sciences. -

Maximum Topological Distances Based Indices As Molecular Descriptors for QSPR
Int. J. Mol. Sci. 2001, 2, 121-132 International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.org/ijms/ Maximum Topological Distances Based Indices as Molecular Descriptors for QSPR. 4. Modeling the Enthalpy of Formation of Hydrocarbons from Elements Andrés Mercader 1, Eduardo A. Castro 1,* and Andrey A. Toropov2 1 CEQUINOR, Departamento de Química, Facultad de Ciencias Exactas, Universidad Nacional de la Plata, C.C. 962, La Plata 1900, Argentina. e-mail: [email protected] 2 Andrey A. Toropov, Vostok Innovation Company, S. Azimstreet 4, 100047 Tashkent, Uzbekistan. e-mail: [email protected] * Author to whom correspondence should be addressed. e-mail: [email protected] Received: 9 April 2001 Accepted: 15 June 2001/ Published: 28 June 2001 Abstract: The enthalpy of formation of a set of 60 hydroarbons is calculated on the basis of topological descriptors defined from the distance and detour matrices within the realm of the QSAR/QSPR theory. Linear and non-linear polynomials fittings are made and results show the need to resort to higher-order regression equations in order to get better concordances between theoretical results and experimental available data. Besides, topological indices computed from maximum order distances seems to yield rather satisfactory predictions of heats of formation for hydrocarbons. Keywords: QSPR theory, Detour matrix, Enthalpy of formation, Hydrocarbons, Topologi- cal indices 1. Introduction Graphs have found considerable employment in several chemistry fields, particularly in modeling molecular structure [1-10]. The applications of graphs to the study of structure-property relationships implies the representation of molecules by selected molecular descriptors, often referred to as topo- logical indices [11,12]. -

Carbene Rearrangements: Intramolecular Interaction of a Triple Bond with a Carbene Center
An Abstract OF THE THESIS OF Jose C. Danino for the degree of Doctor of Philosophy in Chemistry presented on _Dcc, Title: Carbene RearrangementE) Intramolecular Interaction of a Triple Bond with aCarbene Center Redacted for Privacy Abstract approved: Dr. Vetere. Freeman The tosylhydrazones of2-heptanone, 4,4-dimethy1-2- heptanone, 6-heptyn-2-one and 4,4-dimethy1-6-heptyn-2- one were synthesizedand decomposed under a varietyof reaction conditions:' drylithium and sodium salt pyrolyses, sodium methoxide thermolysesin diglyme and photolyses of the lithium salt intetrahydrofuran. The saturated ana- logues 2-heptanone tosylhydrazoneand its 4,4-dimethyl isomer afforded the alkenesarising from 6-hydrogeninser- product distribution in the tion. It was determined that differ- dry salt pyrolyses of2-heptanone tosylhydrazone was ent for the lithiumand the sodium salts. However, the product distribution of thedry sodium salt was verysimilar diglyme to product distributionobtained on thermolysis in explained by a with sodium methoxide. This difference was reaction of lithium bromide(present as an impurity inall compound to the lithium salts)with the intermediate diazo afford an organolithiumintermediate that behaves in a some- what different fashionthan the free carbene.The unsaturated analogues were found to produce a cyclic product in addition to the expected acyclic alkenes arising from 3-hydrogen insertion. By comparison of the acyclic alkene distri- bution obtained in the saturated analogues with those in the unsaturated analogues, it was concluded that at leastsome cyclization was occurring via addition of the diazo moiety to the triple bond. It was determined that the organo- lithium intermediateresulting from lithium bromide cat- alyzed decomposition of the diazo compound was incapable of cyclization. -

Trend Analysis for Atmospheric Hydrocarbon Partitioning Using Continuous Thermodynamics
AUGUST 2005 HARSTAD 2977 Trend Analysis for Atmospheric Hydrocarbon Partitioning Using Continuous Thermodynamics K. HARSTAD Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California (Manuscript received 22 April 2004, in final form 31 January 2005) ABSTRACT The partitioning of atmospheric hydrocarbons into vapor and condensed phases when the species count is large is considered using the formalism of continuous thermodynamics. The vapor saturation pressures and condensate species distribution are parameterized using the species normal boiling temperatures. Qualitative trends in activity coefficient values and phase equilibrium behavior that are relevant to the outer planets and Titan are discussed in terms of a much simplified perspective on these aspects of partitioning. The trends found are generally consistent with those from other published atmospheric model results. 1. Introduction with particular emphasis on extraterrestrial bodies Hydrocarbons (and possibly derivative, i.e., oxidized without oxidizing environments. These models are in species) are commonly present in small amounts in general, very complex, involving partly the kinetics of a planetary atmospheres (e.g., Earth, the outer planets, large number of chemical reactions as well as turbulent Titan), and may attain a relatively wide range of carbon diffusion of chemical species in the atmosphere (Do- number. Abiotic generation is possible by chemical in- brijevic et al. 2003; Lebonnois et al. 2001; Lee et al. 2000; Moses et al. 2000; Raulin and Bruston 1996). De- teraction of CO (or CO2) with H2 (Raulin and Bruston 1996; Zolotov and Shock 1999) and, more effectively, spite the relative complexity of the models in the cited references, a totally complete and precise description of photochemistry of CH4 (Dobrijevic et al.