Integrative Molecular Profiling of Triple Negative Breast Cancers Identifies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

The Utility of Genetic Risk Scores in Predicting the Onset of Stroke March 2021 6
DOT/FAA/AM-21/24 Office of Aerospace Medicine Washington, DC 20591 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke Diana Judith Monroy Rios, M.D1 and Scott J. Nicholson, Ph.D.2 1. KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510 Bogotá D.C. Colombia 2. FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 March 2021 NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents thereof. _________________ This publication and all Office of Aerospace Medicine technical reports are available in full-text from the Civil Aerospace Medical Institute’s publications Web site: (www.faa.gov/go/oamtechreports) Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AM-21/24 4. Title and Subtitle 5. Report Date March 2021 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Diana Judith Monroy Rios M.D1, and Scott J. Nicholson, Ph.D.2 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) 1 KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510, Bogotá D.C. Colombia 11. Contract or Grant No. 2 FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 12. Sponsoring Agency name and Address 13. Type of Report and Period Covered Office of Aerospace Medicine Federal Aviation Administration 800 Independence Ave., S.W. -

Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 As a Centrosomally Localized and Microtubule Associated Protein
cells Article Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 as a Centrosomally Localized and Microtubule Associated Protein 1 1, 1 1 Laura Bergmann , Alexander Lang y , Christoph Bross , Simone Altinoluk-Hambüchen , Iris Fey 1, Nina Overbeck 2, Anja Stefanski 2, Constanze Wiek 3, Andreas Kefalas 1, Patrick Verhülsdonk 1, Christian Mielke 4, Dennis Sohn 5, Kai Stühler 2,6, Helmut Hanenberg 3,7, Reiner U. Jänicke 5, Jürgen Scheller 1, Andreas S. Reichert 8 , Mohammad Reza Ahmadian 1 and Roland P. Piekorz 1,* 1 Institute of Biochemistry and Molecular Biology II, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (L.B.); [email protected] (A.L.); [email protected] (C.B.); [email protected] (S.A.-H.); [email protected] (I.F.); [email protected] (A.K.); [email protected] (P.V.); [email protected] (J.S.); [email protected] (M.R.A.) 2 Molecular Proteomics Laboratory, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (N.O.); [email protected] (A.S.); [email protected] (K.S.) 3 Department of Otolaryngology and Head/Neck Surgery, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (C.W.); [email protected] (H.H.) 4 Institute of Clinical Chemistry and Laboratory Diagnostics, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] -

Iii COMPUTATIONAL APPROACHES for ASSESSING KINOME
COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION A Dissertation Presented to the Faculty of the Weill Cornell Graduate School of Medical Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Charles Joseph Murphy August 2018 iii © 2018 Charles Joseph Murphy ALL RIGHTS RESERVED COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION Charles Joseph Murphy, PhD Cornell University 2018 Protein kinases are a diverse family of about 500 proteins that all share the common ability to catalyze phosphorylation of the side chains of amino acids in proteins. Kinases play a vital role across diverse biological functions including proliferation, differentiation, cell migration, and cell-cycle control. Moreover, they are frequently altered across most cancers types and have been a focus for development of anti- cancer drugs, which has led to the development of 38 approved kinase inhibitors as of 2018. In this thesis, I developed two orthogonal computational approaches for investigating kinase function and deregulation. Starting with data from a large cohort of mouse triple negative breast cancer (TNBC) tumors, I use a combination of whole exome sequencing (WES) and RNA-seq to identify somatic alterations that drive individual tumors. I discovered that a large number of these alterations involve protein kinases and subsequent therapeutic targeting led to tumor regression. For my second approach, I utilized a large peptide library dataset from about 300 kinases. Which kinase phosphorylate which phosphorylation site is determined by both kinase- intrinsic and contextual factors. Peptide library approaches provide kinase-intrinsic amino acid specificity, which I used to predict novel kinase substrates and map out kinase phosphorylation networks. -

Genome-Wide Analysis of Transcriptional Bursting-Induced Noise in Mammalian Cells
bioRxiv preprint doi: https://doi.org/10.1101/736207; this version posted August 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Genome-wide analysis of transcriptional bursting-induced noise in mammalian cells Authors: Hiroshi Ochiai1*, Tetsutaro Hayashi2, Mana Umeda2, Mika Yoshimura2, Akihito Harada3, Yukiko Shimizu4, Kenta Nakano4, Noriko Saitoh5, Hiroshi Kimura6, Zhe Liu7, Takashi Yamamoto1, Tadashi Okamura4,8, Yasuyuki Ohkawa3, Itoshi Nikaido2,9* Affiliations: 1Graduate School of Integrated Sciences for Life, Hiroshima University, Higashi-Hiroshima, Hiroshima, 739-0046, Japan 2Laboratory for Bioinformatics Research, RIKEN BDR, Wako, Saitama, 351-0198, Japan 3Division of Transcriptomics, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Fukuoka, 812-0054, Japan 4Department of Animal Medicine, National Center for Global Health and Medicine (NCGM), Tokyo, 812-0054, Japan 5Division of Cancer Biology, The Cancer Institute of JFCR, Tokyo, 135-8550, Japan 6Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Yokohama, Kanagawa, 226-8503, Japan 7Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA, 20147, USA 8Section of Animal Models, Department of Infectious Diseases, National Center for Global Health and Medicine (NCGM), Tokyo, 812-0054, Japan 9Bioinformatics Course, Master’s/Doctoral Program in Life Science Innovation (T-LSI), School of Integrative and Global Majors (SIGMA), University of Tsukuba, Wako, 351-0198, Japan *Corresponding authors Corresponding authors e-mail addresses Hiroshi Ochiai, [email protected] Itoshi Nikaido, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/736207; this version posted August 15, 2019. -

Sirt1 Is Regulated by Mir-135A and Involved in DNA Damage Repair During Mouse Cellular Reprogramming
www.aging-us.com AGING 2020, Vol. 12, No. 8 Research Paper Sirt1 is regulated by miR-135a and involved in DNA damage repair during mouse cellular reprogramming Andy Chun Hang Chen1,2,*, Qian Peng2,*, Sze Wan Fong1, William Shu Biu Yeung1,2, Yin Lau Lee1,2 1Department of Obstetrics and Gynaecology, The University of Hong Kong, Hong Kong SAR, China 2Shenzhen Key Laboratory of Fertility Regulation, The University of Hong Kong Shenzhen Hospital, Shenzhen, China *Co-first authors Correspondence to: Yin Lau Lee, William Shu Biu Yeung; email: [email protected], [email protected] Keywords: mouse induced pluripotent stem cells, cellular reprogramming, Sirt1, miR-135a, DNA damage repair Received: December 30, 2019 Accepted: March 30, 2020 Published: April 26, 2020 Copyright: Chen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Sirt1 facilitates the reprogramming of mouse somatic cells into induced pluripotent stem cells (iPSCs). It is regulated by micro-RNA and reported to be a target of miR-135a. However, their relationship and roles on cellular reprogramming remain unknown. In this study, we found negative correlations between miR-135a and Sirt1 during mouse embryonic stem cells differentiation and mouse embryonic fibroblasts reprogramming. We further found that the reprogramming efficiency was reduced by the overexpression of miR-135a precursor but induced by the miR-135a inhibitor. Co-immunoprecipitation followed by mass spectrometry identified 21 SIRT1 interacting proteins including KU70 and WRN, which were highly enriched for DNA damage repair. -

Genetic Variant in 3' Untranslated Region of the Mouse Pycard Gene
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 3 Title: 4 Genetic Variant in 3’ Untranslated Region of the Mouse Pycard Gene Regulates Inflammasome 5 Activity 6 Running Title: 7 3’UTR SNP in Pycard regulates inflammasome activity 8 Authors: 9 Brian Ritchey1*, Qimin Hai1*, Juying Han1, John Barnard2, Jonathan D. Smith1,3 10 1Department of Cardiovascular & Metabolic Sciences, Lerner Research Institute, Cleveland Clinic, 11 Cleveland, OH 44195 12 2Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 13 44195 14 3Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine of Case Western 15 Reserve University, Cleveland, OH 44195 16 *, These authors contributed equally to this study. 17 Address correspondence to Jonathan D. Smith: email [email protected]; ORCID ID 0000-0002-0415-386X; 18 mailing address: Cleveland Clinic, Box NC-10, 9500 Euclid Avenue, Cleveland, OH 44195, USA. 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.26.437184; this version posted March 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Quantitative trait locus mapping for interleukin-1 release after inflammasome priming and activation 22 was performed on bone marrow-derived macrophages (BMDM) from an AKRxDBA/2 strain intercross. -

UNIVERSITY of CALIFORNIA RIVERSIDE Investigations Into The
UNIVERSITY OF CALIFORNIA RIVERSIDE Investigations into the Role of TAF1-mediated Phosphorylation in Gene Regulation A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Cell, Molecular and Developmental Biology by Brian James Gadd December 2012 Dissertation Committee: Dr. Xuan Liu, Chairperson Dr. Frank Sauer Dr. Frances M. Sladek Copyright by Brian James Gadd 2012 The Dissertation of Brian James Gadd is approved Committee Chairperson University of California, Riverside Acknowledgments I am thankful to Dr. Liu for her patience and support over the last eight years. I am deeply indebted to my committee members, Dr. Frank Sauer and Dr. Frances Sladek for the insightful comments on my research and this dissertation. Thanks goes out to CMDB, especially Dr. Bachant, Dr. Springer and Kathy Redd for their support. Thanks to all the members of the Liu lab both past and present. A very special thanks to the members of the Sauer lab, including Silvia, Stephane, David, Matt, Stephen, Ninuo, Toby, Josh, Alice, Alex and Flora. You have made all the years here fly by and made them so enjoyable. From the Sladek lab I want to thank Eugene, John, Linh and Karthi. Special thanks go out to all the friends I’ve made over the years here. Chris, Amber, Stephane and David, thank you so much for feeding me, encouraging me and keeping me sane. Thanks to the brothers for all your encouragement and prayers. To any I haven’t mentioned by name, I promise I haven’t forgotten all you’ve done for me during my graduate years. -
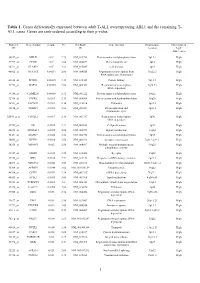
Cases. Genes Are Rank-Ordered According to Their P-Value
Table 1. Genes differentially expressed between adult T-ALL overexpressing ABL1 and the remaining T- ALL cases. Genes are rank-ordered according to their p-value. Probeset Gene symbol p-value FC GenBank Gene function Chromosome Expression in ID ID location “high” ABL1 cases 38847_at MELK <10-4 2.79 NM_014791 Protein amino acid phosphorylation 9p13.2 High 37999_at CPOX <10-4 2.86 NM_000097 Heme biosynthesis 3q12 High 38711_at CLASP2 <10-4 2.33 NM_015097 Cell division 3p23 High 40822_at NFATC3 0.00011 2.06 NM_004555 Regulation of transcription from 16q22.2 High RNA polymerase II promoter 41638_at PPWD1 0.00025 2.13 NM_015342 Protein folding 5q12.3 High 41278_at BAF53 0.00085 2.08 NM_004301 Regulation of transcription, 3q26.33 High DNA-dependent 39388_at CAMK2G 0.00088 2.32 NM_001222 Protein amino acid phosphorylation 10q22 High 32916_at PTPRE 0.0011 2.13 NM_006504 Protein amino acid dephosphorylation 10q26 High 36511_at SACM1L 0.0014 2.16 NM_014016 Unknown 3p21.3 High 38834_at TOPBP1 0.0015 2.55 NM_007027 DNA replication and 3q22.1 High chromosome cycle 33301_g_at CDC2L1 0.0017 2.16 NM_001787 Regulation of transcription, 1p36 High DNA-dependent 32767_at SIL 0.0025 2.23 NM_003035 Cell proliferation 1p32 High 40828_at ARHGEF7 0.0025 2.08 NM_003899 Signal transduction 13q34 High 38431_at MAPK9 0.0026 2.24 NM_002752 Protein amino acid phosphorylation 5q35 High 35663_at NPTX2 0.0028 3.68 NM_002523 Synaptic transmission 7q21.3-q22.1 High 38325_at MINPP1 0.003 2.58 NM_004897 Multiple inositol-polyphosphate 10q23 High phosphatase activity 35747_at -

Anti-PLEKHA1 / TAPP1 Antibody (ARG42988)
Product datasheet [email protected] ARG42988 Package: 100 μl anti-PLEKHA1 / TAPP1 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes PLEKHA1 / TAPP1 Tested Reactivity Hu, Ms, Rat Tested Application IP, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name PLEKHA1 / TAPP1 Antigen Species Human Immunogen Synthetic peptide of Human PLEKHA1 / TAPP1. Conjugation Un-conjugated Alternate Names Pleckstrin homology domain-containing family A member 1; TAPP1; Tandem PH domain-containing protein 1; TAPP-1; PH domain-containing family A member 1 Application Instructions Application table Application Dilution IP 1:20 WB 1:1000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control K562 Calculated Mw 46 kDa Observed Size ~ 46 kDa Properties Form Liquid Purification Affinity purified. Buffer 50 nM Tris-Glycine (pH 7.4), 0.15M NaCl, 0.01% Sodium azide, 40% Glycerol and 0.05% BSA. Preservative 0.01% Sodium azide Stabilizer 40% Glycerol and 0.05% BSA Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/2 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol PLEKHA1 Gene Full Name pleckstrin homology domain containing, family A (phosphoinositide binding specific) member 1 Background This gene encodes a pleckstrin homology domain-containing adapter protein. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -

Structure and Mechanism of the RNA Polymerase II Transcription Machinery
Downloaded from genesdev.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Structure and mechanism of the RNA polymerase II transcription machinery Allison C. Schier and Dylan J. Taatjes Department of Biochemistry, University of Colorado, Boulder, Colorado 80303, USA RNA polymerase II (Pol II) transcribes all protein-coding ingly high resolution, which has rapidly advanced under- genes and many noncoding RNAs in eukaryotic genomes. standing of the molecular basis of Pol II transcription. Although Pol II is a complex, 12-subunit enzyme, it lacks Structural biology continues to transform our under- the ability to initiate transcription and cannot consistent- standing of complex biological processes because it allows ly transcribe through long DNA sequences. To execute visualization of proteins and protein complexes at or near these essential functions, an array of proteins and protein atomic-level resolution. Combined with mutagenesis and complexes interact with Pol II to regulate its activity. In functional assays, structural data can at once establish this review, we detail the structure and mechanism of how enzymes function, justify genetic links to human dis- over a dozen factors that govern Pol II initiation (e.g., ease, and drive drug discovery. In the past few decades, TFIID, TFIIH, and Mediator), pausing, and elongation workhorse techniques such as NMR and X-ray crystallog- (e.g., DSIF, NELF, PAF, and P-TEFb). The structural basis raphy have been complemented by cryoEM, cross-linking for Pol II transcription regulation has advanced rapidly mass spectrometry (CXMS), and other methods. Recent in the past decade, largely due to technological innova- improvements in data collection and imaging technolo- tions in cryoelectron microscopy.