Peripheral Blood Epi-Signature of Claes-Jensen Syndrome Enables Sensitive and Specific Identification of Patients and Healthy Ca
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 As a Centrosomally Localized and Microtubule Associated Protein
cells Article Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 as a Centrosomally Localized and Microtubule Associated Protein 1 1, 1 1 Laura Bergmann , Alexander Lang y , Christoph Bross , Simone Altinoluk-Hambüchen , Iris Fey 1, Nina Overbeck 2, Anja Stefanski 2, Constanze Wiek 3, Andreas Kefalas 1, Patrick Verhülsdonk 1, Christian Mielke 4, Dennis Sohn 5, Kai Stühler 2,6, Helmut Hanenberg 3,7, Reiner U. Jänicke 5, Jürgen Scheller 1, Andreas S. Reichert 8 , Mohammad Reza Ahmadian 1 and Roland P. Piekorz 1,* 1 Institute of Biochemistry and Molecular Biology II, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (L.B.); [email protected] (A.L.); [email protected] (C.B.); [email protected] (S.A.-H.); [email protected] (I.F.); [email protected] (A.K.); [email protected] (P.V.); [email protected] (J.S.); [email protected] (M.R.A.) 2 Molecular Proteomics Laboratory, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (N.O.); [email protected] (A.S.); [email protected] (K.S.) 3 Department of Otolaryngology and Head/Neck Surgery, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (C.W.); [email protected] (H.H.) 4 Institute of Clinical Chemistry and Laboratory Diagnostics, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] -

Iii COMPUTATIONAL APPROACHES for ASSESSING KINOME
COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION A Dissertation Presented to the Faculty of the Weill Cornell Graduate School of Medical Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Charles Joseph Murphy August 2018 iii © 2018 Charles Joseph Murphy ALL RIGHTS RESERVED COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION Charles Joseph Murphy, PhD Cornell University 2018 Protein kinases are a diverse family of about 500 proteins that all share the common ability to catalyze phosphorylation of the side chains of amino acids in proteins. Kinases play a vital role across diverse biological functions including proliferation, differentiation, cell migration, and cell-cycle control. Moreover, they are frequently altered across most cancers types and have been a focus for development of anti- cancer drugs, which has led to the development of 38 approved kinase inhibitors as of 2018. In this thesis, I developed two orthogonal computational approaches for investigating kinase function and deregulation. Starting with data from a large cohort of mouse triple negative breast cancer (TNBC) tumors, I use a combination of whole exome sequencing (WES) and RNA-seq to identify somatic alterations that drive individual tumors. I discovered that a large number of these alterations involve protein kinases and subsequent therapeutic targeting led to tumor regression. For my second approach, I utilized a large peptide library dataset from about 300 kinases. Which kinase phosphorylate which phosphorylation site is determined by both kinase- intrinsic and contextual factors. Peptide library approaches provide kinase-intrinsic amino acid specificity, which I used to predict novel kinase substrates and map out kinase phosphorylation networks. -

Sirt1 Is Regulated by Mir-135A and Involved in DNA Damage Repair During Mouse Cellular Reprogramming
www.aging-us.com AGING 2020, Vol. 12, No. 8 Research Paper Sirt1 is regulated by miR-135a and involved in DNA damage repair during mouse cellular reprogramming Andy Chun Hang Chen1,2,*, Qian Peng2,*, Sze Wan Fong1, William Shu Biu Yeung1,2, Yin Lau Lee1,2 1Department of Obstetrics and Gynaecology, The University of Hong Kong, Hong Kong SAR, China 2Shenzhen Key Laboratory of Fertility Regulation, The University of Hong Kong Shenzhen Hospital, Shenzhen, China *Co-first authors Correspondence to: Yin Lau Lee, William Shu Biu Yeung; email: [email protected], [email protected] Keywords: mouse induced pluripotent stem cells, cellular reprogramming, Sirt1, miR-135a, DNA damage repair Received: December 30, 2019 Accepted: March 30, 2020 Published: April 26, 2020 Copyright: Chen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Sirt1 facilitates the reprogramming of mouse somatic cells into induced pluripotent stem cells (iPSCs). It is regulated by micro-RNA and reported to be a target of miR-135a. However, their relationship and roles on cellular reprogramming remain unknown. In this study, we found negative correlations between miR-135a and Sirt1 during mouse embryonic stem cells differentiation and mouse embryonic fibroblasts reprogramming. We further found that the reprogramming efficiency was reduced by the overexpression of miR-135a precursor but induced by the miR-135a inhibitor. Co-immunoprecipitation followed by mass spectrometry identified 21 SIRT1 interacting proteins including KU70 and WRN, which were highly enriched for DNA damage repair. -

Bioinformatics Analyses of Genomic Imprinting
Bioinformatics Analyses of Genomic Imprinting Dissertation zur Erlangung des Grades des Doktors der Naturwissenschaften der Naturwissenschaftlich-Technischen Fakultät III Chemie, Pharmazie, Bio- und Werkstoffwissenschaften der Universität des Saarlandes von Barbara Hutter Saarbrücken 2009 Tag des Kolloquiums: 08.12.2009 Dekan: Prof. Dr.-Ing. Stefan Diebels Berichterstatter: Prof. Dr. Volkhard Helms Priv.-Doz. Dr. Martina Paulsen Vorsitz: Prof. Dr. Jörn Walter Akad. Mitarbeiter: Dr. Tihamér Geyer Table of contents Summary________________________________________________________________ I Zusammenfassung ________________________________________________________ I Acknowledgements _______________________________________________________II Abbreviations ___________________________________________________________ III Chapter 1 – Introduction __________________________________________________ 1 1.1 Important terms and concepts related to genomic imprinting __________________________ 2 1.2 CpG islands as regulatory elements ______________________________________________ 3 1.3 Differentially methylated regions and imprinting clusters_____________________________ 6 1.4 Reading the imprint __________________________________________________________ 8 1.5 Chromatin marks at imprinted regions___________________________________________ 10 1.6 Roles of repetitive elements ___________________________________________________ 12 1.7 Functional implications of imprinted genes _______________________________________ 14 1.8 Evolution and parental conflict ________________________________________________ -

Congenital Disorders of Glycosylation from a Neurological Perspective
brain sciences Review Congenital Disorders of Glycosylation from a Neurological Perspective Justyna Paprocka 1,* , Aleksandra Jezela-Stanek 2 , Anna Tylki-Szyma´nska 3 and Stephanie Grunewald 4 1 Department of Pediatric Neurology, Faculty of Medical Science in Katowice, Medical University of Silesia, 40-752 Katowice, Poland 2 Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, 01-138 Warsaw, Poland; [email protected] 3 Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, W 04-730 Warsaw, Poland; [email protected] 4 NIHR Biomedical Research Center (BRC), Metabolic Unit, Great Ormond Street Hospital and Institute of Child Health, University College London, London SE1 9RT, UK; [email protected] * Correspondence: [email protected]; Tel.: +48-606-415-888 Abstract: Most plasma proteins, cell membrane proteins and other proteins are glycoproteins with sugar chains attached to the polypeptide-glycans. Glycosylation is the main element of the post- translational transformation of most human proteins. Since glycosylation processes are necessary for many different biological processes, patients present a diverse spectrum of phenotypes and severity of symptoms. The most frequently observed neurological symptoms in congenital disorders of glycosylation (CDG) are: epilepsy, intellectual disability, myopathies, neuropathies and stroke-like episodes. Epilepsy is seen in many CDG subtypes and particularly present in the case of mutations -
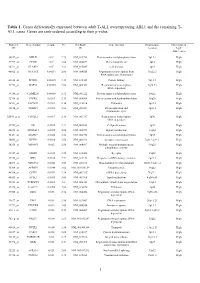
Cases. Genes Are Rank-Ordered According to Their P-Value
Table 1. Genes differentially expressed between adult T-ALL overexpressing ABL1 and the remaining T- ALL cases. Genes are rank-ordered according to their p-value. Probeset Gene symbol p-value FC GenBank Gene function Chromosome Expression in ID ID location “high” ABL1 cases 38847_at MELK <10-4 2.79 NM_014791 Protein amino acid phosphorylation 9p13.2 High 37999_at CPOX <10-4 2.86 NM_000097 Heme biosynthesis 3q12 High 38711_at CLASP2 <10-4 2.33 NM_015097 Cell division 3p23 High 40822_at NFATC3 0.00011 2.06 NM_004555 Regulation of transcription from 16q22.2 High RNA polymerase II promoter 41638_at PPWD1 0.00025 2.13 NM_015342 Protein folding 5q12.3 High 41278_at BAF53 0.00085 2.08 NM_004301 Regulation of transcription, 3q26.33 High DNA-dependent 39388_at CAMK2G 0.00088 2.32 NM_001222 Protein amino acid phosphorylation 10q22 High 32916_at PTPRE 0.0011 2.13 NM_006504 Protein amino acid dephosphorylation 10q26 High 36511_at SACM1L 0.0014 2.16 NM_014016 Unknown 3p21.3 High 38834_at TOPBP1 0.0015 2.55 NM_007027 DNA replication and 3q22.1 High chromosome cycle 33301_g_at CDC2L1 0.0017 2.16 NM_001787 Regulation of transcription, 1p36 High DNA-dependent 32767_at SIL 0.0025 2.23 NM_003035 Cell proliferation 1p32 High 40828_at ARHGEF7 0.0025 2.08 NM_003899 Signal transduction 13q34 High 38431_at MAPK9 0.0026 2.24 NM_002752 Protein amino acid phosphorylation 5q35 High 35663_at NPTX2 0.0028 3.68 NM_002523 Synaptic transmission 7q21.3-q22.1 High 38325_at MINPP1 0.003 2.58 NM_004897 Multiple inositol-polyphosphate 10q23 High phosphatase activity 35747_at -

Current Trends in Gene Recovery Mediated by the CRISPR-Cas System Hyeon-Ki Jang 1, Beomjong Song2,Gue-Hohwang1 and Sangsu Bae 1
Jang et al. Experimental & Molecular Medicine (2020) 52:1016–1027 https://doi.org/10.1038/s12276-020-0466-1 Experimental & Molecular Medicine REVIEW ARTICLE Open Access Current trends in gene recovery mediated by the CRISPR-Cas system Hyeon-Ki Jang 1, Beomjong Song2,Gue-HoHwang1 and Sangsu Bae 1 Abstract The CRISPR-Cas system has undoubtedly revolutionized the genome editing field, enabling targeted gene disruption, regulation, and recovery in a guide RNA-specific manner. In this review, we focus on currently available gene recovery strategies that use CRISPR nucleases, particularly for the treatment of genetic disorders. Through the action of DNA repair mechanisms, CRISPR-mediated DNA cleavage at a genomic target can shift the reading frame to correct abnormal frameshifts, whereas DNA cleavage at two sites, which can induce large deletions or inversions, can correct structural abnormalities in DNA. Homology-mediated or homology-independent gene recovery strategies that require donor DNAs have been developed and widely applied to precisely correct mutated sequences in genes of interest. In contrast to the DNA cleavage-mediated gene correction methods listed above, base-editing tools enable base conversion in the absence of donor DNAs. In addition, CRISPR-associated transposases have been harnessed to generate a targeted knockin, and prime editors have been developed to edit tens of nucleotides in cells. Here, we introduce currently developed gene recovery strategies and discuss the pros and cons of each. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction differs from that of the endogenous gene2. Furthermore, Human genetic disorders, often associated with severe the mutated endogenous gene, which is malfunctional and pathological phenotypes, are caused by genomic aberra- potentially cytotoxic, might still be transcribed. -

A Curated Gene List for Reporting Results of Newborn Genomic Sequencing
© American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE A curated gene list for reporting results of newborn genomic sequencing Ozge Ceyhan-Birsoy, PhD1,2,3, Kalotina Machini, PhD1,2,3, Matthew S. Lebo, PhD1,2,3, Tim W. Yu, MD3,4,5, Pankaj B. Agrawal, MD, MMSC3,4,6, Richard B. Parad, MD, MPH3,7, Ingrid A. Holm, MD, MPH3,4, Amy McGuire, PhD8, Robert C. Green, MD, MPH3,9,10, Alan H. Beggs, PhD3,4, Heidi L. Rehm, PhD1,2,3,10; for the BabySeq Project Purpose: Genomic sequencing (GS) for newborns may enable detec- of newborn GS (nGS), and used our curated list for the first 15 new- tion of conditions for which early knowledge can improve health out- borns sequenced in this project. comes. One of the major challenges hindering its broader application Results: Here, we present our curated list for 1,514 gene–disease is the time it takes to assess the clinical relevance of detected variants associations. Overall, 954 genes met our criteria for return in nGS. and the genes they impact so that disease risk is reported appropri- This reference list eliminated manual assessment for 41% of rare vari- ately. ants identified in 15 newborns. Methods: To facilitate rapid interpretation of GS results in new- Conclusion: Our list provides a resource that can assist in guiding borns, we curated a catalog of genes with putative pediatric relevance the interpretive scope of clinical GS for newborns and potentially for their validity based on the ClinGen clinical validity classification other populations. framework criteria, age of onset, penetrance, and mode of inheri- tance through systematic evaluation of published evidence. -

Associated with Low Dehydrodolichol Diphosphate Synthase (DHDDS) Activity S
Sabry et al. Orphanet Journal of Rare Diseases (2016) 11:84 DOI 10.1186/s13023-016-0468-1 RESEARCH Open Access A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity S. Sabry1,2,3,4, S. Vuillaumier-Barrot1,2,5, E. Mintet1,2, M. Fasseu1,2, V. Valayannopoulos6, D. Héron7,8, N. Dorison8, C. Mignot7,8,9, N. Seta5,10, I. Chantret1,2, T. Dupré1,2,5 and S. E. H. Moore1,2* Abstract Background: Type I congenital disorders of glycosylation (CDG-I) are mostly complex multisystemic diseases associated with hypoglycosylated serum glycoproteins. A subgroup harbour mutations in genes necessary for the biosynthesis of the dolichol-linked oligosaccharide (DLO) precursor that is essential for protein N-glycosylation. Here, our objective was to identify the molecular origins of disease in such a CDG-Ix patient presenting with axial hypotonia, peripheral hypertonia, enlarged liver, micropenis, cryptorchidism and sensorineural deafness associated with hypo glycosylated serum glycoproteins. Results: Targeted sequencing of DNA revealed a splice site mutation in intron 5 and a non-sense mutation in exon 4 of the dehydrodolichol diphosphate synthase gene (DHDDS). Skin biopsy fibroblasts derived from the patient revealed ~20 % residual DHDDS mRNA, ~35 % residual DHDDS activity, reduced dolichol-phosphate, truncated DLO and N-glycans, and an increased ratio of [2-3H]mannose labeled glycoprotein to [2-3H]mannose labeled DLO. Predicted truncated DHDDS transcripts did not complement rer2-deficient yeast. SiRNA-mediated down-regulation of DHDDS in human hepatocellular carcinoma HepG2 cells largely mirrored the biochemical phenotype of cells from the patient. -

MPDU1 (NM 004870) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC200571 MPDU1 (NM_004870) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: MPDU1 (NM_004870) Human Tagged ORF Clone Tag: Myc-DDK Symbol: MPDU1 Synonyms: CDGIF; HBEBP2BPA; Lec35; My008; PP3958; PQLC5; SL15; SLC66A5 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC200571 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCGGCCGAGGCGGACGGACCGCTTAAACGGCTGCTCGTGCCGATTCTTTTACCTGAGAAATGCTACG ACCAACTTTTCGTTCAGTGGGACTTGCTTCACGTCCCCTGCCTCAAGATTCTCCTCAGCAAAGGCCTGGG GCTGGGCATTGTGGCTGGCTCACTTCTAGTAAAGCTGCCCCAGGTGTTTAAAATCCTGGGAGCCAAGAGT GCTGAAGGGTTGAGTCTCCAGTCTGTAATGCTGGAGCTAGTGGCATTGACTGGGACCATGGTCTACAGCA TCACTAACAACTTCCCATTCAGCTCTTGGGGTGAAGCCTTATTCCTGATGCTCCAGACGATCACCATCTG CTTCCTGGTCATGCACTACAGAGGACAGACTGTGAAAGGTGTCGCTTTCCTCGCTTGCTACGGCCTGGTC CTGCTGGTGCTTCTCTCACCTCTGACGCCCTTGACTGTAGTCACCCTGCTCCAGGCCTCCAATGTGCCTG CTGTGGTGGTGGGGAGGCTTCTCCAGGCAGCCACCAACTACCACAACGGGCACACAGGCCAGCTCTCAGC CATCACAGTCTTCCTGCTGTTTGGGGGCTCCCTGGCCCGAATCTTCACTTCCATTCAGGAAACCGGAGAT CCCCTGATGGCTGGGACCTTTGTGGTCTCCTCTCTCTGCAACGGCCTCATCGCCACCCAGCTGCTCTTCT ACTGGAATGCAAAGCCTCCCCACAAGCAGAAAAAGGCGCAG ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Regulation of gene expression programs by serum response factor and megakaryoblastic leukemia 1/2 in macrophages Permalink https://escholarship.org/uc/item/8cc7d0t0 Author Sullivan, Amy Lynn Publication Date 2009 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Regulation of Gene Expression Programs by Serum Response Factor and Megakaryoblastic Leukemia 1/2 in Macrophages A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Amy Lynn Sullivan Committee in charge: Professor Christopher K. Glass, Chair Professor Stephen M. Hedrick Professor Marc R. Montminy Professor Nicholas J. Webster Professor Joseph L. Witztum 2009 Copyright Amy Lynn Sullivan, 2009 All rights reserved. The Dissertation of Amy Lynn Sullivan is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ Chair University of California, San Diego 2009 iii DEDICATION To my husband, Shane, for putting up with me through all of the long hours, last minute late nights, and for not letting me quit no matter how many times my projects fell apart. To my son, Tyler, for always making me smile and for making every day an adventure. To my gifted colleagues, for all of the thought-provoking discussions, technical help and moral support through the roller- coaster ride that has been my graduate career. To my family and friends, for all of your love and support. I couldn’t have done it without you! iv EPIGRAPH If at first you don’t succeed, try, try, again. -

The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagno
International Journal of Molecular Sciences Article The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers Valentina Citro 1, Chiara Cimmaruta 1, Maria Monticelli 1, Guglielmo Riccio 1, Bruno Hay Mele 1,2, Maria Vittoria Cubellis 1,* ID and Giuseppina Andreotti 3 ID 1 Dipartimento di Biologia, Università Federico II, 80126 Napoli, Italy; [email protected] (V.C.); [email protected] (C.C.); [email protected] (M.M.); [email protected] (G.R.); [email protected] (B.H.M.) 2 Dipartimento di Scienze Agrarie ed Agroalimentari, Università Federico II, 80055 Napoli, Italy 3 Istituto di Chimica Biomolecolare—CNR, 80078 Pozzuoli, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-081-679118; Fax: +39-081-679233 Received: 30 May 2018; Accepted: 26 July 2018; Published: 30 July 2018 Abstract: Type I disorders of glycosylation (CDG), the most frequent of which is phosphomannomutase 2 (PMM2-CDG), are a group of diseases causing the incomplete N-glycosylation of proteins. PMM2-CDG is an autosomal recessive disease with a large phenotypic spectrum, and is associated with mutations in the PMM2 gene. The biochemical analysis of mutants does not allow a precise genotype–phenotype correlation for PMM2-CDG. PMM2 is very tolerant to missense and loss of function mutations, suggesting that a partial deficiency of activity might be beneficial under certain circumstances. The patient phenotype might be influenced by variants in other genes associated with the type I disorders of glycosylation in the general population.