Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 As a Centrosomally Localized and Microtubule Associated Protein
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Iii COMPUTATIONAL APPROACHES for ASSESSING KINOME
COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION A Dissertation Presented to the Faculty of the Weill Cornell Graduate School of Medical Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Charles Joseph Murphy August 2018 iii © 2018 Charles Joseph Murphy ALL RIGHTS RESERVED COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION Charles Joseph Murphy, PhD Cornell University 2018 Protein kinases are a diverse family of about 500 proteins that all share the common ability to catalyze phosphorylation of the side chains of amino acids in proteins. Kinases play a vital role across diverse biological functions including proliferation, differentiation, cell migration, and cell-cycle control. Moreover, they are frequently altered across most cancers types and have been a focus for development of anti- cancer drugs, which has led to the development of 38 approved kinase inhibitors as of 2018. In this thesis, I developed two orthogonal computational approaches for investigating kinase function and deregulation. Starting with data from a large cohort of mouse triple negative breast cancer (TNBC) tumors, I use a combination of whole exome sequencing (WES) and RNA-seq to identify somatic alterations that drive individual tumors. I discovered that a large number of these alterations involve protein kinases and subsequent therapeutic targeting led to tumor regression. For my second approach, I utilized a large peptide library dataset from about 300 kinases. Which kinase phosphorylate which phosphorylation site is determined by both kinase- intrinsic and contextual factors. Peptide library approaches provide kinase-intrinsic amino acid specificity, which I used to predict novel kinase substrates and map out kinase phosphorylation networks. -

Barrea Sirtuin.Pdf
Growth Hormone & IGF Research 25 (2015) 28–33 Contents lists available at ScienceDirect Growth Hormone & IGF Research journal homepage: www.elsevier.com/locate/ghir Preliminary data on the relationship between circulating levels of Sirtuin 4, anthropometric and metabolic parameters in obese subjects according to growth hormone/insulin-like growth factor-1 status☆ Silvia Savastano a,⁎,CarolinaDiSommab, Annamaria Colao a, Luigi Barrea c, Francesco Orio d, Carmine Finelli e, Fabrizio Pasanisi a, Franco Contaldo a,GiovanniTarantinof a Dipartimento di Medicina Clinica e Chirurgia, Università Federico II di Napoli, Italy b IRCCS SDN, Napoli, Italy c Coleman & IOS srl, Naples, Italy d Dipartimento di Scienze Motorie e del Benessere Università Parthenope Napoli, Italy e Center of Obesity and Eating Disorders, Stella Maris Mediterraneum Foundation, C/da S. Lucia, Chiaromonte, 80035 Potenza, Italy f Centro Ricerche Oncologiche di Mercogliano, Istituto Nazionale Per Lo Studio e La Cura Dei Tumori “Fondazione Giovanni Pascale”,IRCCS,Italy article info abstract Article history: Background: The main components of GH/insulin-like growth factor (IGF)-1 axis and Sirtuin 4 (Sirt4), highly Received 28 July 2014 expressed in liver and skeletal muscle mitochondria, serve as active regulators of mitochondrial oxidative capac- Received in revised form 20 October 2014 ity with opposite functions. In obesity both GH/IGF-1 status and serum Sirt4 levels, likely mirroring its reduced Accepted 21 October 2014 mitochondrial expression, might be altered. Available online 28 October 2014 Objective: To evaluate the association between circulating levels of Sirt4, body composition, metabolic parame- ters and cardio-metabolic risk profile in obese patients according to their different GH/IGF-1 status. -

Sirt1 Is Regulated by Mir-135A and Involved in DNA Damage Repair During Mouse Cellular Reprogramming
www.aging-us.com AGING 2020, Vol. 12, No. 8 Research Paper Sirt1 is regulated by miR-135a and involved in DNA damage repair during mouse cellular reprogramming Andy Chun Hang Chen1,2,*, Qian Peng2,*, Sze Wan Fong1, William Shu Biu Yeung1,2, Yin Lau Lee1,2 1Department of Obstetrics and Gynaecology, The University of Hong Kong, Hong Kong SAR, China 2Shenzhen Key Laboratory of Fertility Regulation, The University of Hong Kong Shenzhen Hospital, Shenzhen, China *Co-first authors Correspondence to: Yin Lau Lee, William Shu Biu Yeung; email: [email protected], [email protected] Keywords: mouse induced pluripotent stem cells, cellular reprogramming, Sirt1, miR-135a, DNA damage repair Received: December 30, 2019 Accepted: March 30, 2020 Published: April 26, 2020 Copyright: Chen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Sirt1 facilitates the reprogramming of mouse somatic cells into induced pluripotent stem cells (iPSCs). It is regulated by micro-RNA and reported to be a target of miR-135a. However, their relationship and roles on cellular reprogramming remain unknown. In this study, we found negative correlations between miR-135a and Sirt1 during mouse embryonic stem cells differentiation and mouse embryonic fibroblasts reprogramming. We further found that the reprogramming efficiency was reduced by the overexpression of miR-135a precursor but induced by the miR-135a inhibitor. Co-immunoprecipitation followed by mass spectrometry identified 21 SIRT1 interacting proteins including KU70 and WRN, which were highly enriched for DNA damage repair. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -
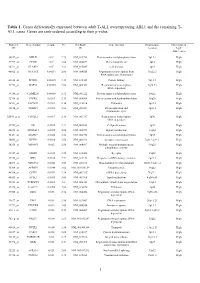
Cases. Genes Are Rank-Ordered According to Their P-Value
Table 1. Genes differentially expressed between adult T-ALL overexpressing ABL1 and the remaining T- ALL cases. Genes are rank-ordered according to their p-value. Probeset Gene symbol p-value FC GenBank Gene function Chromosome Expression in ID ID location “high” ABL1 cases 38847_at MELK <10-4 2.79 NM_014791 Protein amino acid phosphorylation 9p13.2 High 37999_at CPOX <10-4 2.86 NM_000097 Heme biosynthesis 3q12 High 38711_at CLASP2 <10-4 2.33 NM_015097 Cell division 3p23 High 40822_at NFATC3 0.00011 2.06 NM_004555 Regulation of transcription from 16q22.2 High RNA polymerase II promoter 41638_at PPWD1 0.00025 2.13 NM_015342 Protein folding 5q12.3 High 41278_at BAF53 0.00085 2.08 NM_004301 Regulation of transcription, 3q26.33 High DNA-dependent 39388_at CAMK2G 0.00088 2.32 NM_001222 Protein amino acid phosphorylation 10q22 High 32916_at PTPRE 0.0011 2.13 NM_006504 Protein amino acid dephosphorylation 10q26 High 36511_at SACM1L 0.0014 2.16 NM_014016 Unknown 3p21.3 High 38834_at TOPBP1 0.0015 2.55 NM_007027 DNA replication and 3q22.1 High chromosome cycle 33301_g_at CDC2L1 0.0017 2.16 NM_001787 Regulation of transcription, 1p36 High DNA-dependent 32767_at SIL 0.0025 2.23 NM_003035 Cell proliferation 1p32 High 40828_at ARHGEF7 0.0025 2.08 NM_003899 Signal transduction 13q34 High 38431_at MAPK9 0.0026 2.24 NM_002752 Protein amino acid phosphorylation 5q35 High 35663_at NPTX2 0.0028 3.68 NM_002523 Synaptic transmission 7q21.3-q22.1 High 38325_at MINPP1 0.003 2.58 NM_004897 Multiple inositol-polyphosphate 10q23 High phosphatase activity 35747_at -

Charakterisierung Der Interaktion Der Merkelzell-Polyomavirus Kodierten T-Antigene Mit Dem Wirtsfaktor Kap1 Svenja Siebels
Charakterisierung der Interaktion der Merkelzell-Polyomavirus kodierten T-Antigene mit dem Wirtsfaktor Kap1 DISSERTATION zur Erlangung des Doktorgrades (Dr. rer. nat.) an der Fakultät für Mathematik, Informatik und Naturwissenschaften Fachbereich Biologie der Universität Hamburg vorgelegt von Svenja Siebels Hamburg, Juli 2018 Gutachter: Prof. Dr. Nicole Fischer Prof. Dr. Thomas Dobner Disputation: 19. Oktober 2018 Für meine Familie. Zusammenfassung Das Merkelzell-Polyomavirus (MCPyV) ist nachweislich für ca. 80 % aller Merkelzellkarzinome (Merkel cell carcinoma (MCC)) verantwortlich. Das virale Genom ist dabei monoklonal in die DNA der Wirtszelle integriert und trägt zusätzlich charakteristische Mutationen im T-Lokus. Das MCPyV kodiert wie alle Polyomaviren (PyV) die Tumor-Antigene (T-Ag) Large T-Ag und small T-Ag, die transformierende Eigenschaften besitzen. Dennoch sind viele Fragen zur MCC-Entstehung weiterhin ungeklärt. Insbesondere die Ursprungszelle, aus der das MCC hervorgeht, ist ungewiss. Das unvollständige Wissen um den viralen Lebenszyklus sowie die kontroversen Modelle hinsichtlich des Reservoirs des Virus erschweren zusätzlich das Verständnis zur Tumorentstehung. Um das transformierende Potential des MCPyV LT-Ags zu beleuchten, wurden vor Beginn dieser Arbeit neue zelluläre Interaktionspartner des LT-Ags mithilfe von Tandem-Affinitäts-Aufreinigung und anschließender multidimensionaler Protein-Interaktions-Technologie (MudPIT) identifiziert (M. Czech-Sioli, Manuskript in Arbeit). Unter den Kandidaten befand sich das Chromatin-modifizierende Protein, Zellzyklusregulator und Korepressor Kap1 (KRAB-associated protein 1) als putativer Interaktionspartner des LT-Ags. Die Interaktion des LT-Ags, sT-Ags und des verkürzten tLT-Ags (tLT-Ags) mit dem Wirtsfaktor Kap1 wurde in dieser Arbeit mithilfe von Koimmunpräzipitationen in unterschiedlichen Tumorzelllinien bestätigt. Weiterhin wurde die Bindung des LT-Ags an Kap1 auf den N-Terminus des LT-Ags und die RBCC-Domäne von Kap1 eingegrenzt. -

Sirtuins and Diabetes: Optimizing the Sweetness in the Blood Abhinav Kanwal1* and Liston Augustine Dsouza2
Kanwal and Dsouza Translational Medicine Communications (2019) 4:3 Translational Medicine https://doi.org/10.1186/s41231-019-0034-7 Communications REVIEW Open Access Sirtuins and diabetes: optimizing the sweetness in the blood Abhinav Kanwal1* and Liston Augustine Dsouza2 Abstract Diabetes Mellitus (DM) is a chronic disease characterized by elevated levels of glucose in the blood. With time it becomes uncontrollable and invites other complex metabolic diseases. The propensity of the people for this disease is age independent. However, sirtuins, which get activate typically during calorie restriction plays a pivotal role in optimizing effect of blood glucose levels in diabetic patients. Among different sirtuin homologs, some of the sirtuins are known for regulating pathophysiology of diabetic condition. Still the role of other sirtuins in understanding the function and regulatory mechanism in DM is still emerging. In this review, we focused on recent studies which help us to understand about the role of sirtuins and how they regulate the pathophysiology in diabetic condition. Keywords: Sirtuins, Diabetes, Diseases, Mitochondria, Exercise, Deacetylation Background deacetylation status and regulates various factors affect- SIRTUINS are the NAD+ dependent enzymes having an ing disease conditions associated with energy metabol- ability to alter the acetaylation/deacetylation status of ism, aging and oxidative stress [9]. Initially, most of the various proteins present in the body. Mammalian sir- studies on sirtuins were focused on cancer biology but tuins consists of 7 members, Sirt1-Sirt7, that are differ- now the paradigm is shifted to unveil their role in other entiated based on their subcellular localization, substrate disease conditions including diabetes. On the other selectivity etc. -

A Transcriptional Signature of Postmitotic Maintenance in Neural Tissues
Neurobiology of Aging 74 (2019) 147e160 Contents lists available at ScienceDirect Neurobiology of Aging journal homepage: www.elsevier.com/locate/neuaging Postmitotic cell longevityeassociated genes: a transcriptional signature of postmitotic maintenance in neural tissues Atahualpa Castillo-Morales a,b, Jimena Monzón-Sandoval a,b, Araxi O. Urrutia b,c,*, Humberto Gutiérrez a,** a School of Life Sciences, University of Lincoln, Lincoln, UK b Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, UK c Instituto de Ecología, Universidad Nacional Autónoma de México, Ciudad de México, Mexico article info abstract Article history: Different cell types have different postmitotic maintenance requirements. Nerve cells, however, are Received 11 April 2018 unique in this respect as they need to survive and preserve their functional complexity for the entire Received in revised form 3 October 2018 lifetime of the organism, and failure at any level of their supporting mechanisms leads to a wide range of Accepted 11 October 2018 neurodegenerative conditions. Whether these differences across tissues arise from the activation of Available online 19 October 2018 distinct cell typeespecific maintenance mechanisms or the differential activation of a common molecular repertoire is not known. To identify the transcriptional signature of postmitotic cellular longevity (PMCL), Keywords: we compared whole-genome transcriptome data from human tissues ranging in longevity from 120 days Neural maintenance Cell longevity to over 70 years and found a set of 81 genes whose expression levels are closely associated with Transcriptional signature increased cell longevity. Using expression data from 10 independent sources, we found that these genes Functional genomics are more highly coexpressed in longer-living tissues and are enriched in specific biological processes and transcription factor targets compared with randomly selected gene samples. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Regulation of gene expression programs by serum response factor and megakaryoblastic leukemia 1/2 in macrophages Permalink https://escholarship.org/uc/item/8cc7d0t0 Author Sullivan, Amy Lynn Publication Date 2009 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Regulation of Gene Expression Programs by Serum Response Factor and Megakaryoblastic Leukemia 1/2 in Macrophages A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Amy Lynn Sullivan Committee in charge: Professor Christopher K. Glass, Chair Professor Stephen M. Hedrick Professor Marc R. Montminy Professor Nicholas J. Webster Professor Joseph L. Witztum 2009 Copyright Amy Lynn Sullivan, 2009 All rights reserved. The Dissertation of Amy Lynn Sullivan is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ Chair University of California, San Diego 2009 iii DEDICATION To my husband, Shane, for putting up with me through all of the long hours, last minute late nights, and for not letting me quit no matter how many times my projects fell apart. To my son, Tyler, for always making me smile and for making every day an adventure. To my gifted colleagues, for all of the thought-provoking discussions, technical help and moral support through the roller- coaster ride that has been my graduate career. To my family and friends, for all of your love and support. I couldn’t have done it without you! iv EPIGRAPH If at first you don’t succeed, try, try, again. -

"The Genecards Suite: from Gene Data Mining to Disease Genome Sequence Analyses". In: Current Protocols in Bioinformat
The GeneCards Suite: From Gene Data UNIT 1.30 Mining to Disease Genome Sequence Analyses Gil Stelzer,1,5 Naomi Rosen,1,5 Inbar Plaschkes,1,2 Shahar Zimmerman,1 Michal Twik,1 Simon Fishilevich,1 Tsippi Iny Stein,1 Ron Nudel,1 Iris Lieder,2 Yaron Mazor,2 Sergey Kaplan,2 Dvir Dahary,2,4 David Warshawsky,3 Yaron Guan-Golan,3 Asher Kohn,3 Noa Rappaport,1 Marilyn Safran,1 and Doron Lancet1,6 1Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel 2LifeMap Sciences Ltd., Tel Aviv, Israel 3LifeMap Sciences Inc., Marshfield, Massachusetts 4Toldot Genetics Ltd., Hod Hasharon, Israel 5These authors contributed equally to the paper 6Corresponding author GeneCards, the human gene compendium, enables researchers to effectively navigate and inter-relate the wide universe of human genes, diseases, variants, proteins, cells, and biological pathways. Our recently launched Version 4 has a revamped infrastructure facilitating faster data updates, better-targeted data queries, and friendlier user experience. It also provides a stronger foundation for the GeneCards suite of companion databases and analysis tools. Improved data unification includes gene-disease links via MalaCards and merged biological pathways via PathCards, as well as drug information and proteome expression. VarElect, another suite member, is a phenotype prioritizer for next-generation sequencing, leveraging the GeneCards and MalaCards knowledgebase. It au- tomatically infers direct and indirect scored associations between hundreds or even thousands of variant-containing genes and disease phenotype terms. Var- Elect’s capabilities, either independently or within TGex, our comprehensive variant analysis pipeline, help prepare for the challenge of clinical projects that involve thousands of exome/genome NGS analyses. -
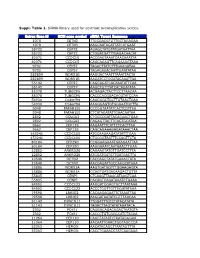
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Rattlesnake Genome Supplemental Materials 1 SUPPLEMENTAL
Rattlesnake Genome Supplemental Materials 1 1 SUPPLEMENTAL MATERIALS 2 Table of Contents 3 1. Supplementary Methods …… 2 4 2. Supplemental Tables ……….. 23 5 3. Supplemental Figures ………. 37 Rattlesnake Genome Supplemental Materials 2 6 1. SUPPLEMENTARY METHODS 7 Prairie Rattlesnake Genome Sequencing and Assembly 8 A male Prairie Rattlesnake (Crotalus viridis viridis) collected from a wild population in Colorado was 9 used to generate the genome sequence. This specimen was collected and humanely euthanized according 10 to University of Northern Colorado Institutional Animal Care and Use Committee protocols 0901C-SM- 11 MLChick-12 and 1302D-SM-S-16. Colorado Parks and Wildlife scientific collecting license 12HP974 12 issued to S.P. Mackessy authorized collection of the animal. Genomic DNA was extracted using a 13 standard Phenol-Chloroform-Isoamyl alcohol extraction from liver tissue that was snap frozen in liquid 14 nitrogen. Multiple short-read sequencing libraries were prepared and sequenced on various platforms, 15 including 50bp single-end and 150bp paired-end reads on an Illumina GAII, 100bp paired-end reads on an 16 Illumina HiSeq, and 300bp paired-end reads on an Illumina MiSeq. Long insert libraries were also 17 constructed by and sequenced on the PacBio platform. Finally, we constructed two sets of mate-pair 18 libraries using an Illumina Nextera Mate Pair kit, with insert sizes of 3-5 kb and 6-8 kb, respectively. 19 These were sequenced on two Illumina HiSeq lanes with 150bp paired-end sequencing reads. Short and 20 long read data were used to assemble the previous genome assembly version CroVir2.0 (NCBI accession 21 SAMN07738522).