Sirt1 Is Regulated by Mir-135A and Involved in DNA Damage Repair During Mouse Cellular Reprogramming
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 As a Centrosomally Localized and Microtubule Associated Protein
cells Article Subcellular Localization and Mitotic Interactome Analyses Identify SIRT4 as a Centrosomally Localized and Microtubule Associated Protein 1 1, 1 1 Laura Bergmann , Alexander Lang y , Christoph Bross , Simone Altinoluk-Hambüchen , Iris Fey 1, Nina Overbeck 2, Anja Stefanski 2, Constanze Wiek 3, Andreas Kefalas 1, Patrick Verhülsdonk 1, Christian Mielke 4, Dennis Sohn 5, Kai Stühler 2,6, Helmut Hanenberg 3,7, Reiner U. Jänicke 5, Jürgen Scheller 1, Andreas S. Reichert 8 , Mohammad Reza Ahmadian 1 and Roland P. Piekorz 1,* 1 Institute of Biochemistry and Molecular Biology II, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (L.B.); [email protected] (A.L.); [email protected] (C.B.); [email protected] (S.A.-H.); [email protected] (I.F.); [email protected] (A.K.); [email protected] (P.V.); [email protected] (J.S.); [email protected] (M.R.A.) 2 Molecular Proteomics Laboratory, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (N.O.); [email protected] (A.S.); [email protected] (K.S.) 3 Department of Otolaryngology and Head/Neck Surgery, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] (C.W.); [email protected] (H.H.) 4 Institute of Clinical Chemistry and Laboratory Diagnostics, Medical Faculty, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany; [email protected] -

Iii COMPUTATIONAL APPROACHES for ASSESSING KINOME
COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION A Dissertation Presented to the Faculty of the Weill Cornell Graduate School of Medical Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Charles Joseph Murphy August 2018 iii © 2018 Charles Joseph Murphy ALL RIGHTS RESERVED COMPUTATIONAL APPROACHES FOR ASSESSING KINOME FUNCTION AND DEREGULATION Charles Joseph Murphy, PhD Cornell University 2018 Protein kinases are a diverse family of about 500 proteins that all share the common ability to catalyze phosphorylation of the side chains of amino acids in proteins. Kinases play a vital role across diverse biological functions including proliferation, differentiation, cell migration, and cell-cycle control. Moreover, they are frequently altered across most cancers types and have been a focus for development of anti- cancer drugs, which has led to the development of 38 approved kinase inhibitors as of 2018. In this thesis, I developed two orthogonal computational approaches for investigating kinase function and deregulation. Starting with data from a large cohort of mouse triple negative breast cancer (TNBC) tumors, I use a combination of whole exome sequencing (WES) and RNA-seq to identify somatic alterations that drive individual tumors. I discovered that a large number of these alterations involve protein kinases and subsequent therapeutic targeting led to tumor regression. For my second approach, I utilized a large peptide library dataset from about 300 kinases. Which kinase phosphorylate which phosphorylation site is determined by both kinase- intrinsic and contextual factors. Peptide library approaches provide kinase-intrinsic amino acid specificity, which I used to predict novel kinase substrates and map out kinase phosphorylation networks. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Variable Penetrance of the 15Q11.2 BP1–BP2 Microduplication in a Family With
bioRxiv preprint doi: https://doi.org/10.1101/095919; this version posted December 22, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Variable penetrance of the 15q11.2 BP1–BP2 microduplication in a family with cognitive and language impairment Antonio Benítez-Burraco1, Montserrat Barcos-Martínez 2,3, Isabel Espejo-Portero2,3, Mª Salud Jiménez-Romero2,4 1. Department of Philology, University of Huelva, Huelva, Spain 2. Maimónides Institute of Biomedical Research, Córdoba, Spain 3. Laboratory of Molecular Genetics, University Hospital “Reina Sofía”, Córdoba, Spain 4. Department of Psychology, University of Córdoba, Córdoba, Spain Corresponding author: Antonio Benítez-Burraco Área de Lengua Española. Departamento de Filología. Facultad de Humanidades. Campus de "El Carmen". Universidad de Huelva. Avda. de las Fuerzas Armadas s/n. 21071-Huelva (España) e-mail: [email protected] Contact details for the other authors: Mª Salud Jiménez-Romero: [email protected] Montserrat Barcos-Martínez: [email protected] Isabel Espejo-Portero: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/095919; this version posted December 22, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 ABSTRACT The 15q11.2 BP1–BP2 region is found duplicated or deleted in people with cognitive, language, and behavioral impairment. Case presentation. We report on a family (the father and three male twin siblings) who presents with a duplication of the 15q11.2 BP1-BP2 region and a variable phenotype: whereas the father and the fraternal twin are normal carriers, the monozygotic twins exhibit severe language and cognitive delay and behavioral disturbances. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -
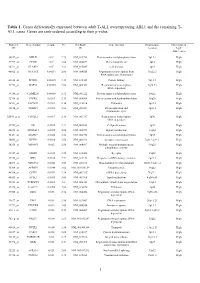
Cases. Genes Are Rank-Ordered According to Their P-Value
Table 1. Genes differentially expressed between adult T-ALL overexpressing ABL1 and the remaining T- ALL cases. Genes are rank-ordered according to their p-value. Probeset Gene symbol p-value FC GenBank Gene function Chromosome Expression in ID ID location “high” ABL1 cases 38847_at MELK <10-4 2.79 NM_014791 Protein amino acid phosphorylation 9p13.2 High 37999_at CPOX <10-4 2.86 NM_000097 Heme biosynthesis 3q12 High 38711_at CLASP2 <10-4 2.33 NM_015097 Cell division 3p23 High 40822_at NFATC3 0.00011 2.06 NM_004555 Regulation of transcription from 16q22.2 High RNA polymerase II promoter 41638_at PPWD1 0.00025 2.13 NM_015342 Protein folding 5q12.3 High 41278_at BAF53 0.00085 2.08 NM_004301 Regulation of transcription, 3q26.33 High DNA-dependent 39388_at CAMK2G 0.00088 2.32 NM_001222 Protein amino acid phosphorylation 10q22 High 32916_at PTPRE 0.0011 2.13 NM_006504 Protein amino acid dephosphorylation 10q26 High 36511_at SACM1L 0.0014 2.16 NM_014016 Unknown 3p21.3 High 38834_at TOPBP1 0.0015 2.55 NM_007027 DNA replication and 3q22.1 High chromosome cycle 33301_g_at CDC2L1 0.0017 2.16 NM_001787 Regulation of transcription, 1p36 High DNA-dependent 32767_at SIL 0.0025 2.23 NM_003035 Cell proliferation 1p32 High 40828_at ARHGEF7 0.0025 2.08 NM_003899 Signal transduction 13q34 High 38431_at MAPK9 0.0026 2.24 NM_002752 Protein amino acid phosphorylation 5q35 High 35663_at NPTX2 0.0028 3.68 NM_002523 Synaptic transmission 7q21.3-q22.1 High 38325_at MINPP1 0.003 2.58 NM_004897 Multiple inositol-polyphosphate 10q23 High phosphatase activity 35747_at -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Gene Expression Analysis of Human Induced Pluripotent Stem Cell
Germain et al. Molecular Autism 2014, 5:44 http://www.molecularautism.com/content/5/1/44 RESEARCH Open Access Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1 Noelle D Germain1, Pin-Fang Chen1, Alex M Plocik1, Heather Glatt-Deeley1, Judith Brown2, James J Fink3, Kaitlyn A Bolduc3, Tiwanna M Robinson3, Eric S Levine3, Lawrence T Reiter4, Brenton R Graveley1,5, Marc Lalande1 and Stormy J Chamberlain1* Abstract Background: Duplications of the chromosome 15q11-q13.1 region are associated with an estimated 1 to 3% of all autism cases, making this copy number variation (CNV) one of the most frequent chromosome abnormalities associated with autism spectrum disorder (ASD). Several genes located within the 15q11-q13.1 duplication region including ubiquitin protein ligase E3A (UBE3A), the gene disrupted in Angelman syndrome (AS), are involved in neural function and may play important roles in the neurobehavioral phenotypes associated with chromosome 15q11-q13.1 duplication (Dup15q) syndrome. Methods: We have generated induced pluripotent stem cell (iPSC) lines from five different individuals containing CNVs of 15q11-q13.1. The iPSC lines were differentiated into mature, functional neurons. Gene expression across the 15q11-q13.1 locus was compared among the five iPSC lines and corresponding iPSC-derived neurons using quantitative reverse transcription PCR (qRT-PCR). Genome-wide gene expression was compared between neurons derived from three iPSC lines using mRNA-Seq. Results: Analysis of 15q11-q13.1 gene expression in neurons derived from Dup15q iPSCs reveals that gene copy number does not consistently predict expression levels in cells with interstitial duplications of 15q11-q13.1. -

A Transcriptional Signature of Postmitotic Maintenance in Neural Tissues
Neurobiology of Aging 74 (2019) 147e160 Contents lists available at ScienceDirect Neurobiology of Aging journal homepage: www.elsevier.com/locate/neuaging Postmitotic cell longevityeassociated genes: a transcriptional signature of postmitotic maintenance in neural tissues Atahualpa Castillo-Morales a,b, Jimena Monzón-Sandoval a,b, Araxi O. Urrutia b,c,*, Humberto Gutiérrez a,** a School of Life Sciences, University of Lincoln, Lincoln, UK b Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Bath, UK c Instituto de Ecología, Universidad Nacional Autónoma de México, Ciudad de México, Mexico article info abstract Article history: Different cell types have different postmitotic maintenance requirements. Nerve cells, however, are Received 11 April 2018 unique in this respect as they need to survive and preserve their functional complexity for the entire Received in revised form 3 October 2018 lifetime of the organism, and failure at any level of their supporting mechanisms leads to a wide range of Accepted 11 October 2018 neurodegenerative conditions. Whether these differences across tissues arise from the activation of Available online 19 October 2018 distinct cell typeespecific maintenance mechanisms or the differential activation of a common molecular repertoire is not known. To identify the transcriptional signature of postmitotic cellular longevity (PMCL), Keywords: we compared whole-genome transcriptome data from human tissues ranging in longevity from 120 days Neural maintenance Cell longevity to over 70 years and found a set of 81 genes whose expression levels are closely associated with Transcriptional signature increased cell longevity. Using expression data from 10 independent sources, we found that these genes Functional genomics are more highly coexpressed in longer-living tissues and are enriched in specific biological processes and transcription factor targets compared with randomly selected gene samples. -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

Downloaded 10 April 2020)
Breeze et al. Genome Medicine (2021) 13:74 https://doi.org/10.1186/s13073-021-00877-z RESEARCH Open Access Epigenome-wide association study of kidney function identifies trans-ethnic and ethnic-specific loci Charles E. Breeze1,2,3* , Anna Batorsky4, Mi Kyeong Lee5, Mindy D. Szeto6, Xiaoguang Xu7, Daniel L. McCartney8, Rong Jiang9, Amit Patki10, Holly J. Kramer11,12, James M. Eales7, Laura Raffield13, Leslie Lange6, Ethan Lange6, Peter Durda14, Yongmei Liu15, Russ P. Tracy14,16, David Van Den Berg17, NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, TOPMed MESA Multi-Omics Working Group, Kathryn L. Evans8, William E. Kraus15,18, Svati Shah15,18, Hermant K. Tiwari10, Lifang Hou19,20, Eric A. Whitsel21,22, Xiao Jiang7, Fadi J. Charchar23,24,25, Andrea A. Baccarelli26, Stephen S. Rich27, Andrew P. Morris28, Marguerite R. Irvin29, Donna K. Arnett30, Elizabeth R. Hauser15,31, Jerome I. Rotter32, Adolfo Correa33, Caroline Hayward34, Steve Horvath35,36, Riccardo E. Marioni8, Maciej Tomaszewski7,37, Stephan Beck2, Sonja I. Berndt1, Stephanie J. London5, Josyf C. Mychaleckyj27 and Nora Franceschini21* Abstract Background: DNA methylation (DNAm) is associated with gene regulation and estimated glomerular filtration rate (eGFR), a measure of kidney function. Decreased eGFR is more common among US Hispanics and African Americans. The causes for this are poorly understood. We aimed to identify trans-ethnic and ethnic-specific differentially methylated positions (DMPs) associated with eGFR using an agnostic, genome-wide approach. Methods: The study included up to 5428 participants from multi-ethnic studies for discovery and 8109 participants for replication. We tested the associations between whole blood DNAm and eGFR using beta values from Illumina 450K or EPIC arrays.