Lawrence Berkeley National Laboratory Recent Work
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Generated by SRI International Pathway Tools Version 25.0, Authors S
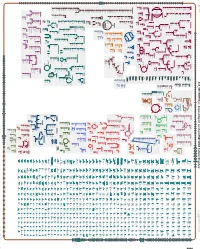
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000238675-HmpCyc: Bacillus smithii 7_3_47FAA Cellular Overview Connections between pathways are omitted for legibility. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Genes for Degradation and Utilization of Uronic Acid-Containing Polysaccharides of a Marine Bacterium Catenovulum Sp
Genes for degradation and utilization of uronic acid-containing polysaccharides of a marine bacterium Catenovulum sp. CCB-QB4 Go Furusawa, Nor Azura Azami and Aik-Hong Teh Centre for Chemical Biology, Universiti Sains Malaysia, Bayan Lepas, Penang, Malaysia ABSTRACT Background. Oligosaccharides from polysaccharides containing uronic acids are known to have many useful bioactivities. Thus, polysaccharide lyases (PLs) and glycoside hydrolases (GHs) involved in producing the oligosaccharides have attracted interest in both medical and industrial settings. The numerous polysaccharide lyases and glycoside hydrolases involved in producing the oligosaccharides were isolated from soil and marine microorganisms. Our previous report demonstrated that an agar-degrading bacterium, Catenovulum sp. CCB-QB4, isolated from a coastal area of Penang, Malaysia, possessed 183 glycoside hydrolases and 43 polysaccharide lyases in the genome. We expected that the strain might degrade and use uronic acid-containing polysaccharides as a carbon source, indicating that the strain has a potential for a source of novel genes for degrading the polysaccharides. Methods. To confirm the expectation, the QB4 cells were cultured in artificial seawater media with uronic acid-containing polysaccharides, namely alginate, pectin (and saturated galacturonate), ulvan, and gellan gum, and the growth was observed. The genes involved in degradation and utilization of uronic acid-containing polysaccharides were explored in the QB4 genome using CAZy analysis and BlastP analysis. Results. The QB4 cells were capable of using these polysaccharides as a carbon source, and especially, the cells exhibited a robust growth in the presence of alginate. 28 PLs and 22 GHs related to the degradation of these polysaccharides were found in Submitted 5 August 2020 the QB4 genome based on the CAZy database. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

High-Quality-Draft Genome Sequence of the Fermenting Bacterium Anaerobium Acetethylicum Type Strain Glubs11t (DSM 29698)
Lawrence Berkeley National Laboratory Recent Work Title High-quality-draft genome sequence of the fermenting bacterium Anaerobium acetethylicum type strain GluBS11T (DSM 29698). Permalink https://escholarship.org/uc/item/2b22m9pw Journal Standards in genomic sciences, 12(1) ISSN 1944-3277 Authors Patil, Yogita Müller, Nicolai Schink, Bernhard et al. Publication Date 2017 DOI 10.1186/s40793-017-0236-4 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Patil et al. Standards in Genomic Sciences (2017) 12:24 DOI 10.1186/s40793-017-0236-4 SHORT GENOME REPORT Open Access High-quality-draft genome sequence of the fermenting bacterium Anaerobium acetethylicum type strain GluBS11T (DSM 29698) Yogita Patil1, Nicolai Müller1*, Bernhard Schink1*, William B. Whitman3, Marcel Huntemann4, Alicia Clum4, Manoj Pillay4, Krishnaveni Palaniappan4, Neha Varghese4, Natalia Mikhailova4, Dimitrios Stamatis4, T. B. K. Reddy4, Chris Daum4, Nicole Shapiro4, Natalia Ivanova4, Nikos Kyrpides4, Tanja Woyke4 and Madan Junghare1,2* Abstract Anaerobium acetethylicum strain GluBS11T belongs to the family Lachnospiraceae within the order Clostridiales.Itisa Gram-positive, non-motile and strictly anaerobic bacterium isolated from biogas slurry that was originally enriched with gluconate as carbon source (Patil, et al., Int J Syst Evol Microbiol 65:3289-3296, 2015). Here we describe the draft genome sequence of strain GluBS11T and provide a detailed insight into its physiological and metabolic features. The draft genome sequence generated 4,609,043 bp, distributed among 105 scaffolds assembled using the SPAdes genome assembler method. It comprises in total 4,132 genes, of which 4,008 were predicted to be protein coding genes, 124 RNA genes and 867 pseudogenes. -

Invasive Escherichia Colito Exposure to Bile Salts
www.nature.com/scientificreports OPEN Metabolic adaptation of adherent- invasive Escherichia coli to exposure to bile salts Received: 7 August 2018 Julien Delmas 1,2, Lucie Gibold1,2, Tiphanie Faïs1,2, Sylvine Batista1, Martin Leremboure4, Accepted: 13 December 2018 Clara Sinel3, Emilie Vazeille2,5, Vincent Cattoir3, Anthony Buisson2,5, Nicolas Barnich2,6, Published: xx xx xxxx Guillaume Dalmasso 2 & Richard Bonnet1,2 The adherent-invasive Escherichia coli (AIEC), which colonize the ileal mucosa of Crohn’s disease patients, adhere to intestinal epithelial cells, invade them and exacerbate intestinal infammation. The high nutrient competition between the commensal microbiota and AIEC pathobiont requires the latter to occupy their own metabolic niches to survive and proliferate within the gut. In this study, a global RNA sequencing of AIEC strain LF82 has been used to observe the impact of bile salts on the expression of metabolic genes. The results showed a global up-regulation of genes involved in degradation and a down-regulation of those implicated in biosynthesis. The main up-regulated degradation pathways were ethanolamine, 1,2-propanediol and citrate utilization, as well as the methyl-citrate pathway. Our study reveals that ethanolamine utilization bestows a competitive advantage of AIEC strains that are metabolically capable of its degradation in the presence of bile salts. We observed that bile salts activated secondary metabolism pathways that communicate to provide an energy beneft to AIEC. Bile salts may be used by AIEC as an environmental signal to promote their colonization. Te adherent-invasive Escherichia coli (AIEC) pathogroup was initially characterized in isolates from the ileal mucosa of Crohn’s disease (CD) patients1–5. -

Kendall High School and the Western New York Genetics in Research and Health Care Partnership
18 A Annotation of the Listeria monocytogenes Genome from DNA Coordinates 1450..2513 and 5886..7337 Alycia A. Davenport*, Bridget K. Miller*, and Kathryn S. Hoppe Kendall High School and the Western New York Genetics in Research and Health Care Partnership Abstract LM5923_0009: The ini tial propo sed product of thi s gene by GENI-ACT was Listeria monocytogenesisprin cipally found in pathogenic ca ses offood Modules ofthe GEN I-ACT (http://w w w .geni-act.org/) were used to hypothetical protein. The top BL AST hits for thi s gene were, poisoning. This organism which cause s Listeriosis, is one of the complete Listeria monocytogene s genome annotation. The however, Rhamnulokinase . The BL AS T hits had signi fi cant and leading causes of death offoodborne pathogens. The bacterium can modules are described below: spr ead from the si te of infe ction in the intestines to the nervous system scor e s and E-value s. The cor r ec t annotation being Modules Activities Questions Investigated Rhamnulokinase isalso suppor ted byTIGRFAM and PFA M hitsfor to the placenta in pregnantindividuals. The purpose of this study was Module 1- Basic Information DNA Coordinates and What is the sequence of my to identify and explain different coding regions of the genome of Module Sequence, Protein Sequence gene and protein? Where is this gene. It is thus proposed tha t LM5923_0009 should be it located in the genome? Listeria monocytogenes. The tw o gene lo ci studied were Listeria annotated asRhamnulokinase .Rhamnulokina se (EC2 .7.1 .5) isan monocytogene 5923_0009 and 5923_0004. -

Modeling and Computational Prediction of Metabolic Channelling
MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING by Christopher Morran Sanford A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Christopher Morran Sanford 2009 Abstract MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING Master of Science 2009 Christopher Morran Sanford Graduate Department of Molecular Genetics University of Toronto Metabolic channelling occurs when two enzymes that act on a common substrate pass that intermediate directly from one active site to the next without allowing it to diffuse into the surrounding aqueous medium. In this study, properties of channelling are investigated through the use of computational models and cell simulation tools. The effects of enzyme kinetics and thermodynamics on channelling are explored with the emphasis on validating the hypothesized roles of metabolic channelling in living cells. These simulations identify situations in which channelling can induce acceleration of reaction velocities and reduction in the free concentration of intermediate metabolites. Databases of biological information, including metabolic, thermodynamic, toxicity, inhibitory, gene fusion and physical protein interaction data are used to predict examples of potentially channelled enzyme pairs. The predictions are used both to support the hypothesized evolutionary motivations for channelling, and to propose potential enzyme interactions that may be worthy of future investigation. ii Acknowledgements I wish to thank my supervisor Dr. John Parkinson for the guidance he has provided during my time spent in his lab, as well as for his extensive help in the writing of this thesis. I am grateful for the advice of my committee members, Prof. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Genomic, Transcriptomic and Enzymatic Insight Into Lignocellulolytic System of a Plant Pathogen Dickeya Sp
biomolecules Article Genomic, Transcriptomic and Enzymatic Insight into Lignocellulolytic System of a Plant Pathogen Dickeya sp. WS52 to Digest Sweet Pepper and Tomato Stalk 1, 2, 3, 2 2 Ying-Jie Yang y, Wei Lin y, Raghvendra Pratap Singh * , Qian Xu , Zhihou Chen , Yuan Yuan 1 , Ping Zou 1, Yiqiang Li 1 and Chengsheng Zhang 1,* 1 Marine Agriculture Research Center, Tobacco Research Institute of Chinese Academy of Agricultural Sciences, Qingdao 266101, China; [email protected] (Y.-J.Y.); [email protected] (Y.Y.); [email protected] (P.Z.); [email protected] (Y.L.) 2 Tobacco Research Institute of Nanping, Nanping, Fujian 353000, China; [email protected] (W.L.); [email protected] (Q.X.); [email protected] (Z.C.) 3 Department of Research & Development, Biotechnology, Uttaranchal University, Dehradun 248007, India * Correspondence: [email protected] (R.P.S.); [email protected] (C.Z.) These authors contributed equally to this work. y Received: 31 October 2019; Accepted: 18 November 2019; Published: 20 November 2019 Abstract: Dickeya sp., a plant pathogen, causing soft rot with strong pectin degradation capacity was taken for the comprehensive analysis of its corresponding biomass degradative system, which has not been analyzed yet. Whole genome sequence analysis of the isolated soft-rotten plant pathogen Dickeya sp. WS52, revealed various coding genes which involved in vegetable stalk degradation-related properties. A total of 122 genes were found to be encoded for putative carbohydrate-active enzymes (CAZy) in Dickeya sp. WS52. The number of pectin degradation-related genes, was higher than that of cellulolytic bacteria as well as other Dickeya spp.