Downloaded Genomes And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomics 98 (2011) 370–375
Genomics 98 (2011) 370–375 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Whole-genome comparison clarifies close phylogenetic relationships between the phyla Dictyoglomi and Thermotogae Hiromi Nishida a,⁎, Teruhiko Beppu b, Kenji Ueda b a Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan b Life Science Research Center, College of Bioresource Sciences, Nihon University, Fujisawa, Japan article info abstract Article history: The anaerobic thermophilic bacterial genus Dictyoglomus is characterized by the ability to produce useful Received 2 June 2011 enzymes such as amylase, mannanase, and xylanase. Despite the significance, the phylogenetic position of Accepted 1 August 2011 Dictyoglomus has not yet been clarified, since it exhibits ambiguous phylogenetic positions in a single gene Available online 7 August 2011 sequence comparison-based analysis. The number of substitutions at the diverging point of Dictyoglomus is insufficient to show the relationships in a single gene comparison-based analysis. Hence, we studied its Keywords: evolutionary trait based on whole-genome comparison. Both gene content and orthologous protein sequence Whole-genome comparison Dictyoglomus comparisons indicated that Dictyoglomus is most closely related to the phylum Thermotogae and it forms a Bacterial systematics monophyletic group with Coprothermobacter proteolyticus (a constituent of the phylum Firmicutes) and Coprothermobacter proteolyticus Thermotogae. Our findings indicate that C. proteolyticus does not belong to the phylum Firmicutes and that the Thermotogae phylum Dictyoglomi is not closely related to either the phylum Firmicutes or Synergistetes but to the phylum Thermotogae. © 2011 Elsevier Inc. -

Modification of the Campylobacter Jejuni Flagellin Glycan by the Product of the Cj1295 Homopolymeric-Tract-Containing Gene
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Microbiology (2010), 156, 1953–1962 DOI 10.1099/mic.0.038091-0 Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene Paul Hitchen,1,2 Joanna Brzostek,1 Maria Panico,1 Jonathan A. Butler,3 Howard R. Morris,1,4 Anne Dell1 and Dennis Linton3 Correspondence 1Division of Molecular Biosciences, Faculty of Natural Science, Imperial College, Dennis Linton London SW7 2AY, UK [email protected] 2Centre for Integrative Systems Biology at Imperial College, Faculty of Natural Science, Imperial College, London SW7 2AY, UK 3Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, UK 4M-SCAN Ltd, Wokingham, Berkshire RG41 2TZ, UK The Campylobacter jejuni flagellin protein is O-glycosylated with structural analogues of the nine- carbon sugar pseudaminic acid. The most common modifications in the C. jejuni 81-176 strain are the 5,7-di-N-acetylated derivative (Pse5Ac7Ac) and an acetamidino-substituted version (Pse5Am7Ac). Other structures detected include O-acetylated and N-acetylglutamine- substituted derivatives (Pse5Am7Ac8OAc and Pse5Am7Ac8GlnNAc, respectively). Recently, a derivative of pseudaminic acid modified with a di-O-methylglyceroyl group was detected in C. jejuni NCTC 11168 strain. The gene products required for Pse5Ac7Ac biosynthesis have been characterized, but those genes involved in generating other structures have not. We have demonstrated that the mobility of the NCTC 11168 flagellin protein in SDS-PAGE gels can vary spontaneously and we investigated the role of single nucleotide repeats or homopolymeric-tract- containing genes from the flagellin glycosylation locus in this process. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000238675-HmpCyc: Bacillus smithii 7_3_47FAA Cellular Overview Connections between pathways are omitted for legibility. -

Campylobacter Is a Genus of Gram-Negative, Microaerophilic, Motile, Rod-Shaped Enteric Bacteria
For Vets General Information • Campylobacter is a genus of Gram-negative, microaerophilic, motile, rod-shaped enteric bacteria. • It is the most commonly diagnosed cause of bacterial diarrhea in people in the developed world. • There are several significant species of Campylobacter found in both people and animals. The most common species that cause disease in humans are C. jejuni subsp. jejuni (often simply called C. jejuni) and C. coli, which account for up to 95% of all human cases - C. jejuni is most often associated with chickens, but it is found in pets as well. The most common species found in dogs and cats is C. upsaliensis, which uncommonly infects humans. Cats can also commonly carry C. helveticus, but this species’ role in human disease (if any) remains unclear. • Campylobacter is an important cause of disease in humans. Disease in animals is much less common, but the bacterium is often found in healthy pets. When illness occurs, the most common sign is diarrhea. • Campylobacter infection can spread beyond the gastrointestinal tract, resulting in severe, even life-threatening systemic illness, particularly in young, elderly or immunocompromised individuals. • The risk of transmission of Campylobacter between animals and people can be reduced by increasing awareness of the means of transmission and some common-sense infection control measures. Prevalence & Risk Factors Humans • Campylobacteriosis is one of the most commonly diagnosed causes of bacterial enteric illness in humans worldwide. In Canada, annual disease rates have been estimated to be 26.7 cases/100 000 person-years, but because diarrheal diseases are typically under-reported, the true incidence is likely much higher. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title What we can deduce about metabolism in the moderate halophile chromohalobacter salexigens from its genomic sequence Permalink https://escholarship.org/uc/item/1df4q5m5 Authors Csonka, Laszlo N. O'Connor, Kathleen Larimer, Frank et al. Publication Date 2005 eScholarship.org Powered by the California Digital Library University of California 1 Biodata of Laszlo N. Csonka, author of “What We Can Deduce about Metabolism in the Moderate Halophile Chromohalobacter salexigens from its Genomic Sequence” Dr. Laszlo Csonka is a professor in the Department of Biological Sciences at Purdue University. He received his Ph. D. in 1975 at Harvard Medical School in the laboratory of Dr. Dan Fraenkel, studying the pathways of NADPH formation in Escherichia coli. His main research interests are the analysis of the response to osmotic stress in bacteria and plants, the connection between osmotic adaptation and thermotolerance, and carbon- source metabolism in bacteria. E-mail: [email protected] 2 3 WHAT WE CAN DEDUCE ABOUT METABOLISM IN THE MODERATE HALOPHILE CHROMOHALOBACTER SALEXIGENS FROM ITS GENOMIC SEQUENCE LASZLO N. CSONKA1, KATHLEEN O’CONNOR1, FRANK LARIMER2, PAUL RICHARDSON3, ALLA LAPIDUS3, ADAM D. EWING4, BRADLEY W. GOODNER4 and AHARON OREN5 1Department of Biological Sciences, Purdue University, West Lafayette IN 47907-1392, USA; 2Genome Analysis and Systems Modeling, Life Sciences Division, Oak Ridge National Laboratory, Oak Ridge TN 37831, USA; 3DOE Joint Genome Institute, Walnut Creek CA 94598, USA; 4Department of Biology, Hiram College, Hiram OH 44234, USA; 5The Institute of Life Science, and the Moshe Shilo Minerva Center for Marine Biogeochemistry, The Hebrew University of Jerusalem, 91904, Israel 1. -

Arcobacter, Campylobacter, and Helicobacter in Reptiles
fmicb-10-01086 May 28, 2019 Time: 15:12 # 1 REVIEW published: 15 May 2019 doi: 10.3389/fmicb.2019.01086 Living in Cold Blood: Arcobacter, Campylobacter, and Helicobacter in Reptiles Maarten J. Gilbert1,2*, Birgitta Duim1,3, Aldert L. Zomer1,3 and Jaap A. Wagenaar1,3,4 1 Department of Infectious Diseases and Immunology, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands, 2 Reptile, Amphibian and Fish Conservation Netherlands, Nijmegen, Netherlands, 3 WHO Collaborating Center for Campylobacter/OIE Reference Laboratory for Campylobacteriosis, Utrecht, Netherlands, 4 Wageningen Bioveterinary Research, Lelystad, Netherlands Species of the Epsilonproteobacteria genera Arcobacter, Campylobacter, and Helicobacter are commonly associated with vertebrate hosts and some are considered significant pathogens. Vertebrate-associated Epsilonproteobacteria are often considered to be largely confined to endothermic mammals and birds. Recent studies have shown that ectothermic reptiles display a distinct and largely unique Epsilonproteobacteria community, including taxa which can cause disease in humans. Several Arcobacter taxa are widespread amongst reptiles and often show a broad host range. Reptiles carry a large diversity of unique and novel Helicobacter taxa, which Edited by: John R. Battista, apparently evolved in an ectothermic host. Some species, such as Campylobacter Louisiana State University, fetus, display a distinct intraspecies host dichotomy, with genetically divergent lineages United States occurring either in mammals or reptiles. These taxa can provide valuable insights in Reviewed by: Heriberto Fernandez, host adaptation and co-evolution between symbiont and host. Here, we present an Austral University of Chile, Chile overview of the biodiversity, ecology, epidemiology, and evolution of reptile-associated Zuowei Wu, Epsilonproteobacteria from a broader vertebrate host perspective. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Campylobacter and Rotavirus Co-Infection in Diarrheal Children in a Referral Children Hospital in Nepal
Campylobacter and Rotavirus co-infection in diarrheal children in a referral children hospital in Nepal Vishnu Bhattarai Tribhuvan University Institute of Science and Technology Saroj Sharma Kantipur City College Komal Raj Rijal ( [email protected] ) Tribhuvan University https://orcid.org/0000-0001-6281-8236 Megha Raj Banjara Tribhuvan University Institute of Science and Technology Research article Keywords: Campylobacter , Rotavirus, co-infection, diarrhea, children Posted Date: August 23rd, 2019 DOI: https://doi.org/10.21203/rs.2.13512/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on February 13th, 2020. See the published version at https://doi.org/10.1186/s12887-020-1966-9. Page 1/13 Abstract Diarrhea, although easily curable, is a global cause of death for a million children every year. Rotavirus and Campylobacter are the most common etiological agents of diarrhea in children under 5 years of age. However, in Nepal, these causative agents are not routinely examined for the diagnosis and treatment. The objective of this study was to determine Campylobacter co-infection associated with Rotaviral diarrhea in children less than 5 years of age. A cross-sectional study was conducted at Kanti Children's Hospital (KCH), Kathmandu, Nepal from November 2017 to April 2018. A total of 303 stool specimens from diarrheal children were processed to detect Rotavirus using rapid Rotavirus Ag test kit, and Campylobacter by microscopy, culture and biochemical tests. Antibiotic susceptibility test of Campylobacter isolates was performed according to European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines 2015. -
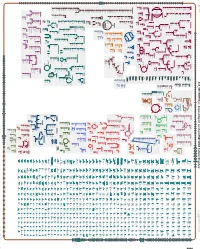
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Cosr Regulation of Perr Transcription for the Control of Oxidative Stress Defense in Campylobacter Jejuni
microorganisms Communication CosR Regulation of perR Transcription for the Control of Oxidative Stress Defense in Campylobacter jejuni Myungseo Park 1,†, Sunyoung Hwang 2,3,†,‡, Sangryeol Ryu 2,3,4,* and Byeonghwa Jeon 1,* 1 Division of Environmental Health Sciences, School of Public Health, University of Minnesota, Minneapolis, MN 55455, USA; [email protected] 2 Department of Food and Animal Biotechnology, Research Institute for Agriculture and Life Sciences, Seoul National University, Seoul 08826, Korea; [email protected] 3 Department of Agricultural Biotechnology, Research Institute for Agriculture and Life Sciences, Seoul National University, Seoul 08826, Korea 4 Center for Food Bioconvergence, Seoul National University, Seoul 08826, Korea * Correspondence: [email protected] (S.R.); [email protected] (B.J.) † The authors equally contributed. ‡ Current address: Food Microbiology Division/Food Safety Evaluation Department, National Institute of Food and Drug Safety Evaluation, Osong 28159, Korea. Abstract: Oxidative stress resistance is an important mechanism to sustain the viability of oxygen- sensitive microaerophilic Campylobacter jejuni. In C. jejuni, gene expression associated with oxidative stress defense is modulated by PerR (peroxide response regulator) and CosR (Campylobacter oxidative stress regulator). Iron also plays an important role in the regulation of oxidative stress, as high iron concentrations reduce the transcription of perR. However, little is known about how iron affects the transcription of cosR. The level of cosR transcription was increased when the defined media MEMα (Minimum Essential Medium) was supplemented with ferrous (Fe2+) and ferric (Fe3+) iron and the Citation: Park, M.; Hwang, S.; Ryu, Mueller–Hinton (MH) media was treated with an iron chelator, indicating that iron upregulates S.; Jeon, B. -

Genes for Degradation and Utilization of Uronic Acid-Containing Polysaccharides of a Marine Bacterium Catenovulum Sp
Genes for degradation and utilization of uronic acid-containing polysaccharides of a marine bacterium Catenovulum sp. CCB-QB4 Go Furusawa, Nor Azura Azami and Aik-Hong Teh Centre for Chemical Biology, Universiti Sains Malaysia, Bayan Lepas, Penang, Malaysia ABSTRACT Background. Oligosaccharides from polysaccharides containing uronic acids are known to have many useful bioactivities. Thus, polysaccharide lyases (PLs) and glycoside hydrolases (GHs) involved in producing the oligosaccharides have attracted interest in both medical and industrial settings. The numerous polysaccharide lyases and glycoside hydrolases involved in producing the oligosaccharides were isolated from soil and marine microorganisms. Our previous report demonstrated that an agar-degrading bacterium, Catenovulum sp. CCB-QB4, isolated from a coastal area of Penang, Malaysia, possessed 183 glycoside hydrolases and 43 polysaccharide lyases in the genome. We expected that the strain might degrade and use uronic acid-containing polysaccharides as a carbon source, indicating that the strain has a potential for a source of novel genes for degrading the polysaccharides. Methods. To confirm the expectation, the QB4 cells were cultured in artificial seawater media with uronic acid-containing polysaccharides, namely alginate, pectin (and saturated galacturonate), ulvan, and gellan gum, and the growth was observed. The genes involved in degradation and utilization of uronic acid-containing polysaccharides were explored in the QB4 genome using CAZy analysis and BlastP analysis. Results. The QB4 cells were capable of using these polysaccharides as a carbon source, and especially, the cells exhibited a robust growth in the presence of alginate. 28 PLs and 22 GHs related to the degradation of these polysaccharides were found in Submitted 5 August 2020 the QB4 genome based on the CAZy database. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0