Proteomic Analysis of Methanonatronarchaeum Thermophilum AMET1, a Representative of a Putative New Class of Euryarchaeota, “Methanonatronarchaeia”
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Review Article: Inhibition of Methanogenic Archaea by Statins As a Targeted Management Strategy for Constipation and Related Disorders
Alimentary Pharmacology and Therapeutics Review article: inhibition of methanogenic archaea by statins as a targeted management strategy for constipation and related disorders K. Gottlieb*, V. Wacher*, J. Sliman* & M. Pimentel† *Synthetic Biologics, Inc., Rockville, SUMMARY MD, USA. † Gastroenterology, Cedars-Sinai Background Medical Center, Los Angeles, CA, USA. Observational studies show a strong association between delayed intestinal transit and the production of methane. Experimental data suggest a direct inhibitory activity of methane on the colonic and ileal smooth muscle and Correspondence to: a possible role for methane as a gasotransmitter. Archaea are the only con- Dr K. Gottlieb, Synthetic Biologics, fi Inc., 9605 Medical Center Drive, rmed biological sources of methane in nature and Methanobrevibacter Rockville, MD 20850, USA. smithii is the predominant methanogen in the human intestine. E-mail: [email protected] Aim To review the biosynthesis and composition of archaeal cell membranes, Publication data archaeal methanogenesis and the mechanism of action of statins in this context. Submitted 8 September 2015 First decision 29 September 2015 Methods Resubmitted 7 October 2015 Narrative review of the literature. Resubmitted 20 October 2015 Accepted 20 October 2015 Results EV Pub Online 11 November 2015 Statins can inhibit archaeal cell membrane biosynthesis without affecting This uncommissioned review article was bacterial numbers as demonstrated in livestock and humans. This opens subject to full peer-review. the possibility of a therapeutic intervention that targets a specific aetiologi- cal factor of constipation while protecting the intestinal microbiome. While it is generally believed that statins inhibit methane production via their effect on cell membrane biosynthesis, mediated by inhibition of the HMG- CoA reductase, there is accumulating evidence for an alternative or addi- tional mechanism of action where statins inhibit methanogenesis directly. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Structure and Function of the Archaeal Response Regulator Chey
Structure and function of the archaeal response PNAS PLUS regulator CheY Tessa E. F. Quaxa, Florian Altegoerb, Fernando Rossia, Zhengqun Lia, Marta Rodriguez-Francoc, Florian Krausd, Gert Bangeb,1, and Sonja-Verena Albersa,1 aMolecular Biology of Archaea, Faculty of Biology, University of Freiburg, 79104 Freiburg, Germany; bLandes-Offensive zur Entwicklung Wissenschaftlich- ökonomischer Exzellenz Center for Synthetic Microbiology & Faculty of Chemistry, Philipps-University-Marburg, 35043 Marburg, Germany; cCell Biology, Faculty of Biology, University of Freiburg, 79104 Freiburg, Germany; and dFaculty of Chemistry, Philipps-University-Marburg, 35043 Marburg, Germany Edited by Norman R. Pace, University of Colorado at Boulder, Boulder, CO, and approved December 13, 2017 (received for review October 2, 2017) Motility is a central feature of many microorganisms and provides display different swimming mechanisms. Counterclockwise ro- an efficient strategy to respond to environmental changes. Bacteria tation results in smooth swimming in well-characterized peritri- and archaea have developed fundamentally different rotary motors chously flagellated bacteria such as Escherichia coli, whereas enabling their motility, termed flagellum and archaellum, respec- rotation in the opposite direction results in tumbling. In contrast, tively. Bacterial motility along chemical gradients, called chemo- in other bacteria (i.e., Vibrio alginolyticus) and haloarchaea, taxis, critically relies on the response regulator CheY, which, when clockwise rotation results -

Bradymonabacteria, a Novel Bacterial Predator Group with Versatile
Mu et al. Microbiome (2020) 8:126 https://doi.org/10.1186/s40168-020-00902-0 RESEARCH Open Access Bradymonabacteria, a novel bacterial predator group with versatile survival strategies in saline environments Da-Shuai Mu1,2, Shuo Wang2, Qi-Yun Liang2, Zhao-Zhong Du2, Renmao Tian3, Yang Ouyang3, Xin-Peng Wang2, Aifen Zhou3, Ya Gong1,2, Guan-Jun Chen1,2, Joy Van Nostrand3, Yunfeng Yang4, Jizhong Zhou3,4 and Zong-Jun Du1,2* Abstract Background: Bacterial predation is an important selective force in microbial community structure and dynamics. However, only a limited number of predatory bacteria have been reported, and their predatory strategies and evolutionary adaptations remain elusive. We recently isolated a novel group of bacterial predators, Bradymonabacteria, representative of the novel order Bradymonadales in δ-Proteobacteria. Compared with those of other bacterial predators (e.g., Myxococcales and Bdellovibrionales), the predatory and living strategies of Bradymonadales are still largely unknown. Results: Based on individual coculture of Bradymonabacteria with 281 prey bacteria, Bradymonabacteria preyed on diverse bacteria but had a high preference for Bacteroidetes. Genomic analysis of 13 recently sequenced Bradymonabacteria indicated that these bacteria had conspicuous metabolic deficiencies, but they could synthesize many polymers, such as polyphosphate and polyhydroxyalkanoates. Dual transcriptome analysis of cocultures of Bradymonabacteria and prey suggested a potential contact-dependent predation mechanism. Comparative genomic analysis with 24 other bacterial predators indicated that Bradymonabacteria had different predatory and living strategies. Furthermore, we identified Bradymonadales from 1552 publicly available 16S rRNA amplicon sequencing samples, indicating that Bradymonadales was widely distributed and highly abundant in saline environments. Phylogenetic analysis showed that there may be six subgroups in this order; each subgroup occupied a different habitat. -

Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China Di Wu Northeast Forestry University Caihong Zhao Northeast Forestry University Hui Bai Forestry Science Research Institute of Heilongjiang Province Fujuan Feng Northeast Forestry University Xin Sui Heilongjiang University Guangyu Sun ( [email protected] ) Northeast Forestry University Research article Keywords: Wetlands, Methanogens, Community diversity, Indicator species, Methanogenic metabolic patterns Posted Date: August 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-54821/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Soil methanogenic microorganisms are one of the primary methane-producing microbes in wetlands. However, we still poorly understand the community characteristic and metabolic patterns of these microorganisms according to vegetation type and seasonal changes. Therefore, to better elucidate the effects of the vegetation type and seasonal factors on the methanogenic community structure and metabolic patterns, we detected the characteristics of the soil methanogenic mcrA gene from three types of natural wetlands in different seasons in the Xiaoxing'an Mountain region, China. Result: The results indicated that the distribution of Methanobacteriaceae (hydrogenotrophic methanogens) was higher in winter, while Methanosarcinaceae and Methanosaetaceae accounted for a higher proportion in summer. Hydrogenotrophic methanogenesis was the dominant trophic pattern in each wetland. The results of principal coordinate analysis and cluster analysis showed that the vegetation type considerably inuenced the methanogenic community composition. The methanogenic community structure in the Betula platyphylla – Larix gmelinii wetland was relatively different from the structure of the other two wetland types. -

Motile Ghosts of the Halophilic Archaeon, Haloferax Volcanii
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.08.899351; this version posted May 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Motile ghosts of the halophilic archaeon, 2 Haloferax volcanii 3 Yoshiaki Kinosita1,2,¶,*, Nagisa Mikami2, Zhengqun Li2, Frank Braun2, Tessa EF. Quax2, 4 Chris van der Does2, Robert Ishmukhametov1, Sonja-Verena Albers2 & Richard M. Berry1 5 1Department of Physics, University of Oxford, Park load OX1 3PU, Oxford, UK 6 2Institute for Biology II, University of Freiburg, Schaenzle strasse 1, 79104 Freiburg, 7 Germany 8 ¶Present address: Molecular Physiology Laboratory, RIKEN, Japan 9 *Correspondence should be addressed to [email protected] 10 Author Contributions: 11 Y.K. and R.M.B designed the research. Y.K. performed all experiments and 12 obtained all data; N.M. helped genetics, biochemistry, and preparation of figures; 13 Z.L, F.B., T.EF.Q., C.v.d.D and S.-V. A. helped genetics; R.I helped the ghost 14 experiments; N.M. and R.M.B helped microscope measurements; Y.K., and 15 R.M.B. wrote the paper. 16 17 18 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.01.08.899351; this version posted May 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
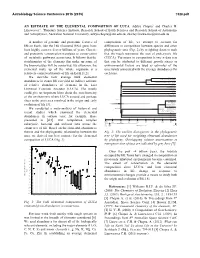
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio
International Journal of New Technology and Research (IJNTR) ISSN:2454-4116, Volume-2, Issue-9, September 2016 Pages 81-86 Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio S. Matsuda, T. Ohtsuki* Anaerobic digestion is a conventional method for biomass Abstract— Acclimation of microbial communities, aiming to utilization [8]. Despite the fact that a great deal of research methane production from grass as a sole substrate at extremely for methane production from grass has been conducted high carbon-to-nitrogen (C/N) ratio, was conducted. In a series especially in recent years, almost processes need supply of of experiments with various sizes of added grass, two microbial communities showing high methane production were obtained abundant sludge source such as sewage and manure [9, 10]. with powdered grass. In the two microbial communities Many studies indicated that the optimal carbon-to-nitrogen designated NR and RP, Bacteroidia including genera (C/N) ratio in methane fermentation were 25-30, and the C/N Bacteroides, Dysgonomonas, Proteiniphilum, and Alistipes were ratio higher than 40 are not generally suitable [11]. The C/N detected as dominant members in eubacteria. It was also shown ration of raw grass shows a wide range; wild grassy weed is at that Methanomicrobia and Methanobacteria including genera relatively high (>59) while lawn grass is at low (<29) [12]. Methanomassiliicoccus and Methanobacterium were found as dominant members in methanogen. It is noteworthy that Therefore, it is difficult to conduct methane fermentation nitrogen fixation were observed both in NR and RP, suggesting with wild grass-only, also being due to less degradability of that insufficiency of nitrogen sources would be complemented lignocellulose [13]. -

Viruses of Hyperthermophilic Archaea: Entry and Egress from the Host Cell
Viruses of hyperthermophilic archaea : entry and egress from the host cell Emmanuelle Quemin To cite this version: Emmanuelle Quemin. Viruses of hyperthermophilic archaea : entry and egress from the host cell. Microbiology and Parasitology. Université Pierre et Marie Curie - Paris VI, 2015. English. NNT : 2015PA066329. tel-01374196 HAL Id: tel-01374196 https://tel.archives-ouvertes.fr/tel-01374196 Submitted on 30 Sep 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Université Pierre et Marie Curie – Paris VI Unité de Biologie Moléculaire du Gène chez les Extrêmophiles Ecole doctorale Complexité du Vivant ED515 Département de Microbiologie - Institut Pasteur 7, quai Saint-Bernard, case 32 25, rue du Dr. Roux 75252 Paris Cedex 05 75015 Paris THESE DE DOCTORAT DE L’UNIVERSITE PIERRE ET MARIE CURIE Spécialité : Microbiologie Pour obtenir le grade de DOCTEUR DE L’UNIVERSITE PIERRE ET MARIE CURIE VIRUSES OF HYPERTHERMOPHILIC ARCHAEA: ENTRY INTO AND EGRESS FROM THE HOST CELL Présentée par M. Emmanuelle Quemin Soutenue le 28 Septembre 2015 devant le jury composé de : Prof. Guennadi Sezonov Président du jury Prof. Christa Schleper Rapporteur de thèse Dr. Paulo Tavares Rapporteur de thèse Dr. -

Structure of the Archaellar Motor and Associated Cytoplasmic Cone In
bioRxiv preprint first posted online Feb. 13, 2017; doi: http://dx.doi.org/10.1101/108209. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder. It is made available under a CC-BY-NC-ND 4.0 International license. 1 Structure of the archaellar motor and associated cytoplasmic cone in 2 Thermococcus kodakaraensis 3 4 Ariane Briegel1,2, Catherine M. Oikonomou1, Yi-Wei Chang1, Andreas Kjær1,3, Audrey N. 5 Huang1, Ki Woo Kim4, Debnath Ghosal1, Robert P. Gunsalus5, and Grant J. Jensen1,6,* 6 7 8 9 1 Division of Biology and Biological Engineering, California Institute of Technology, 1200 E. 10 California Blvd., Pasadena, CA 91125 11 2 Current: Institute of Biology, Leiden University, Sylviusweg 72, 2333 BE Leiden, Netherlands 12 3 Current address: University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark 13 4 School of Ecology and Environmental System, Kyungpook National University, Sangju 37224, 14 South Korea 15 5 Department of Microbiology, Immunology and Molecular Genetics, the Molecular Biology 16 Institute, University of California, Los Angeles, 609 Charles E. Young Dr. S., Los Angeles, CA 17 90095 18 6 Howard Hughes Medical Institute, 1200 E. California Blvd., Pasadena, CA 91125 19 * Correspondence: [email protected] 20 21 Keywords: electron cryotomography, cryo-EM, archaea, archaella, flagella, T4P, motility, 22 Thermococcus kodakaraensis, Thermococcus kodakarensis 23 1 bioRxiv preprint first posted online Feb. 13, 2017; doi: http://dx.doi.org/10.1101/108209. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder. It is made available under a CC-BY-NC-ND 4.0 International license. -

1 Development of Prediction Models of Methane Production by Sheep
Development of Prediction Models of Methane Production by Sheep and Cows Using Rumen Microbiota Data Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Boyang Zhang Graduate Program in Animal Sciences The Ohio State University 2018 Master's Examination Committee: Dr. Zhongtang Yu, Advisor Dr. Moraes, Co-advisor Dr. Firkins 1 Copyrighted by Boyang Zhang 2018 2 Abstract Methane emission from the rumen leads to approximately 10 % loss of the ingested energy, at the conversion of digestible to metabolizable energy. Besides research on mitigation of methane emission, much research interests have also been gravitated towards development of prediction models of methane emissions from livestock because the global warming is reducing agriculture productivity (Johnson and Johnson, 1995). An accurate prediction of enteric methane production from cattle and sheep can assist in balancing the increased livestock production with subsequent environmental impacts. Methane is an inevitable byproduct of the microbial fermentation processes in the rumen, and certain ruminal microbes have direct impacts to methane production (Morgavi et al., 2010). Thus, we hypothesized that the inclusion of individual microbial groups as predictor variables could improve the robustness and accuracy of prediction models. However, inclusion of microbial variables into prediction models can result in overfitting. Machine-learning algorithms can automatically select the key explanatory predictors, and Linear Mixed Models can provide a framework to predict random effects describing between animal variations. We proposed three novel frameworks for subset selections of microbial variables (MV) and one framework for generalized linear mixed models (GLMM) using L1-penalization (GLMMLASSO) selection with cross-validations (CV) ii to address the overfitting problems and to develop parsimonious prediction models of methane production.