Novel Potent and Selective DPP-4 Inhibitors: Design, Synthesis and Molecular Docking Study of Dihydropyrimidine Phthalimide Hybrids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CDR Clinical Review Report for Soliqua
CADTH COMMON DRUG REVIEW Clinical Review Report Insulin glargine and lixisenatide injection (Soliqua) (Sanofi-Aventis) Indication: adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus inadequately controlled on basal insulin (less than 60 units daily) alone or in combination with metformin. Service Line: CADTH Common Drug Review Version: Final (with redactions) Publication Date: January 2019 Report Length: 118 Pages Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services. While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. -
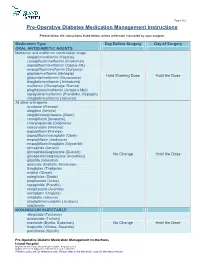
Pre-Operative Diabetes Medication Management Instructions
Page 1 of 2 Pre-Operative Diabetes Medication Management Instructions Please follow the instructions listed below unless otherwise instructed by your surgeon Medication Type Day Before Surgery Day of Surgery ORAL ANTIDIABETIC AGENTS Metformin and metformin combination drugs alogliptin/metformin (Kazano) canagliflozin/metformin (Invokamet) dapagliflozin/metformin (Xigduo XR) empagliflozin/metformin (Synjardy) glipizide/metformin (Metaglip) Hold Evening Dose Hold the Dose glyburide/metformin (Glucovance) linagliptin/metformin (Jentadueto) metformin (Glucophage, Riomet) pioglitazone/metformin (Actoplus Met) repaglidine/metformin (PrandiMet, Repaglin) sitagliptin/metformin (Janumet) All other oral agents acarbose (Precose) alogliptin (Nesina) alogliptin/pioglitazone (Oseni) canagliflozin (Invokana) chlorpropamide (Diabinese) colesevelam (Welchol) dapagliflozin (Farxiga) dapagliflozin/saxagliptin (Qtern) empagliflozin (Jardiance) empagliflozin/linagliptin (Glyxambi) glimepiride (Amaryl) glimepiride/pioglitazone (Duetact) No Change Hold the Dose glimepiride/rosiglitazone (Avandaryl) glipizide (Glucotrol) glyburide (DiaBeta, Micronase) linagliptan (Tradjenta) miglitol (Glyset) nateglinide (Starlix) pioglitazone (Actos) repaglinide (Prandin) rosiglitazone (Avandia) saxagliptin (Onglyza) sitagliptin (Januvia) sitagliptin/simvastatin (Juvisync) tolazamide NON-INSULIN INJECTABLE albiglutide (Tanzeum) dulaglutide (Trulicity) exenatide (Byetta, Bydureon) No Change Hold the Dose liraglutide (Victoza, Saxenda) pramlintide (Symlin) Pre-Operative Diabetic -

Antidiabetic Drugs in NAFLD: the Accomplishment of Two Goals at Once?
pharmaceuticals Review Antidiabetic Drugs in NAFLD: The Accomplishment of Two Goals at Once? Matteo Tacelli , Ciro Celsa , Bianca Magro, Aurora Giannetti, Grazia Pennisi, Federica Spatola and Salvatore Petta * Sezione di Gastroenterologia e Epatologia, DiBiMIS, University of Palermo, 90127 Palermo, Italy; [email protected] (M.T.); [email protected] (C.C.); [email protected] (B.M.); [email protected] (A.G.); [email protected] (G.P.); [email protected] (F.S.) * Correspondence: [email protected], Tel.: +39-091-655-2170; Fax: +39-091-655-2156 Received: 17 October 2018; Accepted: 3 November 2018; Published: 8 November 2018 Abstract: Non-Alcoholic Fatty Liver Disease (NAFLD) is the most common cause of chronic liver disease in Western countries, accounting for 20–30% of general population and reaching a prevalence of 55% in patients with type 2 diabetes mellitus (T2DM). Insulin resistance plays a key role in pathogenic mechanisms of NAFLD. Many drugs have been tested but no medications have yet been approved. Antidiabetic drugs could have a role in the progression reduction of the disease. The aim of this review is to summarize evidence on efficacy and safety of antidiabetic drugs in patients with NAFLD. Metformin, a biguanide, is the most frequently used drug in the treatment of T2DM. To date 15 randomized controlled trials (RCTs) and four meta-analysis on the use of metformin in NAFLD are available. No significant improvement in histological liver fibrosis was shown, but it can be useful in the treatment of co-factors of NAFLD, like body weight, transaminase or cholesterol levels, and HbA1c levels. -

OSENI (Alogliptin and Pioglitazone) Tablets II May Increase Risk
HIGHLIGHTS OF PRESCRIBING INFORMATION -----------------------WARNINGS AND PRECAUTIONS--------------------- These highlights do not include all the information needed to use • Congestive heart failure: Fluid retention may occur and can OSENI safely and effectively. See full prescribing information for exacerbate or lead to congestive heart failure. Combination use OSENI. with insulin and use in congestive heart failure NYHA Class I and OSENI (alogliptin and pioglitazone) tablets II may increase risk. Monitor patients for signs and symptoms. Initial U.S. Approval: 2013 (5.1) • Acute pancreatitis: There have been postmarketing reports of WARNING: CONGESTIVE HEART FAILURE acute pancreatitis. If pancreatitis is suspected, promptly See full prescribing information for complete boxed warning discontinue OSENI. (5.2) • Thiazolidinediones, including pioglitazone, cause or • Hypersensitivity: There have been postmarketing reports of exacerbate congestive heart failure in some patients. (5.1) serious hypersensitivity reactions in patients treated with alogliptin • After initiation of OSENI and after dose increases, monitor such as anaphylaxis, angioedema and severe cutaneous adverse patients carefully for signs and symptoms of heart failure reactions. In such cases, promptly discontinue OSENI, assess for (e.g., excessive, rapid weight gain, dyspnea and/or other potential causes, institute appropriate monitoring and edema). If heart failure develops, it should be managed treatment and initiate alternative treatment for diabetes. (5.3) according to current standards of care and • Hepatic effects: Postmarketing reports of hepatic failure, discontinuation or dose reduction of pioglitazone in OSENI sometimes fatal. Causality cannot be excluded. If liver injury is must be considered. detected, promptly interrupt OSENI and assess patient for • OSENI is not recommended in patients with symptomatic probable cause, then treat cause if possible, to resolution or heart failure. -

NESINA (Alogliptin) Tablets, for Oral Use • Heart Failure: Consider the Risks and Benefits of NESINA Prior to Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION ------------------------WARNINGS AND PRECAUTIONS---------------------- These highlights do not include all the information needed to use • Acute pancreatitis: There have been postmarketing reports of NESINA safely and effectively. See full prescribing information for acute pancreatitis. If pancreatitis is suspected, promptly NESINA. discontinue NESINA. (5.1) NESINA (alogliptin) tablets, for oral use • Heart failure: Consider the risks and benefits of NESINA prior to Initial U.S. Approval: 2013 initiating treatment in patients at risk for heart failure. If heart failure develops, evaluate and manage according to current ---------------------------RECENT MAJOR CHANGES-------------------------- standards of care and consider discontinuation of NESINA (5.2). Indications and Usage (1.1) 4/2016 • Hypersensitivity: There have been postmarketing reports of Dosage and Administration serious hypersensitivity reactions in patients treated with NESINA Patients with Renal Impairment (2.2) 4/2016 such as anaphylaxis, angioedema and severe cutaneous adverse Warnings and Precautions reactions, including Stevens-Johnson syndrome. In such cases, Pancreatitis (5.1) 4/2016 promptly discontinue NESINA, assess for other potential causes, Heart Failure (5.2) 4/2016 institute appropriate monitoring and treatment and initiate Hepatic Effects (5.4) 4/2016 alternative treatment for diabetes. (5.3) Bullous Pemphigoid (5.7) 12/2016 • Hepatic effects: Postmarketing reports of hepatic failure, ----------------------------INDICATIONS AND USAGE--------------------------- sometimes fatal. Causality cannot be excluded. If liver injury is NESINA is a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an detected, promptly interrupt NESINA and assess patient for adjunct to diet and exercise to improve glycemic control in adults with probable cause, then treat cause if possible, to resolution or type 2 diabetes mellitus. -

Diabetes Recommendations and Tier Coverage Chart
DIABETES RECOMMENDATIONS AND TIER COVERAGE CHART The American Diabetes Association guidelines for 2020, recommend metformin as the preferred initial treatment for type 2 diabetes (T2DM) along with weight management and physical activity. In patients who have established ASVD or at high risk, CKD, or HF, a SGLT2i or GLP-1 receptor with proven efficacy is recommended independent of A1C. • ASCVD dominates: o GLP-1RA with proven CVD benefit (dulaglutide, liraglutide, injectable semaglutide) OR o SGLT2i with proven CVD benefit (canagliflozin, empagliflozin) if adequate eGFR • HF or CKD dominates: o SGLT2i with evidence of reducing HF and/or CKD progression (empagliflozin, canagliflozin, dapagliflozin) if adequate eGFR OR o If SGLT2i intolerant/contraindicated or eGFR is inadequate, then GLP-1RA with proven CVD benefit In individuals without established cardiovascular disease, pharmacological treatment should be patient-centered taking into account side-effects, cost, impact on weight, risk of hypoglycemia, and other patient preferences. For more detailed information regarding ADA recommendations for pharmacological agents to treat T2DM click here. The following chart is a list of oral and injectable diabetes medications listed by class with their respective A1C reduction and insurance coverage and/or coverage requirements for BCBS, HPHC, Tufts, TMP, and MassHealth. Tufts Medicare Medications BCBSMA HPHC Tufts Preferred MassHealth Biguanides A1C reduction: 1-1.5% metformin Tier 1 Tier 1;2 Tier 1 Tier 1 Covered Glucoghage (metformin) NC NC NC;Tier -

Comparison of Glimepiride, Alogliptin and Alogliptin+Pioglitazone Combination in Poorly Controlled Type 2 Diabetic Patients ( Protocol: Takeda ALO-IIT)
Takeda_ALO-IIT_Ver 3.3 date: 29/Dec/2016 Comparison of glimepiride, alogliptin and alogliptin+pioglitazone combination in poorly controlled type 2 diabetic patients ( Protocol: Takeda_ALO-IIT) Version No: 3.3 date:29/Dec/2016 Principal Investigator’s Affiliation: Seoul National University Bundang Hospital 1 Takeda_ALO-IIT_Ver 3.3 date: 29/Dec/2016 Principal Investigator’s Name: Sung Hee Choi Research Outline Comparison of glimepiride, alogliptin and alogliptin+pioglitazone combination in Title of Research poorly controlled type 2 diabetic patients Principal Investigator Professor Sung Hee Choi Institution Supporting Takeda Pharmaceuticals Korea Co. Ltd. Research Expenses The primary objective is to compare the change in HbA1c in week 24 in 3 treatment groups: the glimepiride monotherapy treatment group; the alogliptin monotherapy treatment group; the alogliptin - pioglitazone combination therapy treatment group. - The secondary objective is to compare the change in HbA1c in week 12 and fasting plasma glucose (FPG) in week 12 and 24 in the following 3 treatment groups over the course of 3 months (at the Baseline, in Week 12): (the glimepiride Research Objective monotherapy treatment group, the alogliptin monotherapy treatment group, and the alogliptin - pioglitazone combination therapy treatment group). - Also, the change in parameters of glycemic variability assessed by CGM will be investigated. - Also, for a 6-month period, the average change in the lipid profile will be compared (Baseline, Week 12, Week 24). · This trial is a three-armed, open label, random assignment trial. · The research subjects are patients who are first starting their treatment or patients who have failed with the metformin treatment and are changing their medication. -

Initial Combination Therapy with Canagliflozin Plus Metformin Versus Each Component As Monotherapy for Drug-Naïve Type 2 Diabe
Diabetes Care Volume 39, March 2016 353 Julio Rosenstock,1 Leonard Chuck,2 Initial Combination Therapy With Manuel Gonzalez-Ortiz,´ 3 Kate Merton,4 CLIN CARE/EDUCATION/NUTRITION/PSYCHOSOCIAL Jagriti Craig,4 George Capuano,4 and Canagliflozin Plus Metformin Rong Qiu4 Versus Each Component as Monotherapy for Drug-Na¨ıve Type 2 Diabetes Diabetes Care 2016;39:353–362 | DOI: 10.2337/dc15-1736 OBJECTIVE This study assessed the efficacy/safety of canagliflozin (CANA), a sodium–glucose cotransporter 2 (SGLT2) inhibitor, plus metformin extended-release (MET) initial therapy in drug-na¨ıve type 2 diabetes. RESEARCH DESIGN AND METHODS This 26-week, double-blind, phase 3 study randomized 1,186 patients to CANA 100 mg (CANA100)/MET, CANA 300 mg (CANA300)/MET, CANA100, CANA300, or MET. Primary end point was change in HbA1c at week 26 for combinations versus monotherapies. Secondary end points included noninferiority in HbA1c lowering with CANA monotherapy versus MET; changes in fasting plasma glucose, body weight, and blood pressure; and proportion of patients achieving HbA1c <7.0% (<53 mmol/mol). RESULTS From mean baseline HbA1c of 8.8% (73 mmol/mol), CANA100/MET and CANA300/ MET significantly lowered HbA1c versus MET (median dose, 2,000 mg/day) by –1.77%, –1.78%, and –1.30% (–19.3, –19.5, and –14.2 mmol/mol; differences of 1Dallas Diabetes and Endocrine Center at Medi- 2 – – – P cal City, Dallas, TX 0.46% and 0.48% [ 5.0 and 5.2 mmol/mol]; = 0.001) and versus CANA100 2 – – – – Diablo Clinical Research, Walnut Creek, CA and CANA300 by 1.37% and 1.42% ( 15.0 and 15.5 mmol/mol; differences of 3Institute of Experimental and Clinical Therapeu- –0.40% and –0.36% [–4.4 and –3.9 mmol/mol]; P = 0.001). -

Combination Use of Insulin and Incretins in Type 2 Diabetes
Canadian Agency for Agence canadienne Drugs and Technologies des médicaments et des in Health technologies de la santé CADTH Optimal Use Report Volume 3, Issue 1C Combination Use of Insulin and July 2013 Incretins in Type 2 Diabetes Supporting Informed Decisions This report is prepared by the Canadian Agency for Drugs and Technologies in Health (CADTH). The report contains a comprehensive review of the existing public literature, studies, materials, and other information and documentation (collectively the “source documentation”) available to CADTH at the time of report preparation. The information in this report is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. The information in this report should not be used as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process, nor is it intended to replace professional medical advice. While CADTH has taken care in the preparation of this document to ensure that its contents are accurate, complete, and up to date as of the date of publication, CADTH does not make any guarantee to that effect. CADTH is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in the source documentation. CADTH is not responsible for any errors or omissions or injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the information in this document or in any of the source documentation. -

ANTI-HYPERGLYCEMIC DIABETES AGENTS in T2DM: Color Outcomes Comparison Summary Table
ANTI-HYPERGLYCEMIC DIABETES AGENTS in T2DM: Outcomes Comparison Summary Table L Regier BSP BA, M LeBras PharmD, T Trischuk PharmD, J Bareham BSP, L Lu BSP © www.RxFiles.ca Aug 2021 Drug Class Sulfonylureas TZDs Meglitinides DPP4 Inhibitors GLP1 Agonists *** SGLT2 Inhibitors *** Insulin in T2DM Generic Metformin Gliclazide Glyburide Pioglitazone Rosiglitazone Acarbose Repaglinide Saxagliptin ONGLYZA Liraglutide VICTOZA Empagliflozin JARDIANCE Intensity: Intensity: BRAND (MF) DIAMICRON DIABETA ACTOS, g AVANDIA GLUCOBAY GLUCONORM Sitagliptin JANUVIA Exenatide BYETTA, BYDUREON Canagliflozin INVOKANA Less More GLUCOPHAGE Dulaglutide TRULICITY Dapagliflozin FORXIGA, FARXIGA Alogliptin NESINA (e.g. NPH (Multiple daily GLUCOTROL D/C STEGLATRO Glipizide Nateglinide Semaglutide OZEMPIC, RYBELSUS (PO) Ertugliflozin Linagliptin TRAJENTA HS + MF) doses) STARLIX D/C SPREAD-DIMCAD] Lixisenatide ADLYXINE; ALBIGLUTIDE D/C SAVOR-TIMI 53, EMPA-REG, CANVAS, CREDENCE, T2DM: UKPDS-33,80; ADVANCE, Major trials to UKPDS- ProACTIVE ACE 33,34,80 UKPDS- Meta-analysis. TECOS, EXAMINE LEADER, EXSCEL, FREEDOM CVO, DECLARE, VERTIS-CV (2020), ACCORD, VADT, ORIGIN, DEVOTE support ADVANCE (Prevention (ADOPT; 33,80 Ferwana M. Meta- RECORD interim, PROLOGUE, REWIND, SUSTAIN-6, PIONEER-6, DAPA-HF, DAPA-CKD (2020), T1DM: DCCT/EDIC findings/ analysis 2013. trial: Stop- - some use in (ADOPT) ADOPT, DREAM CARMELINA, EMPEROR-Reduced & -Preserved (Also Boussageon et al. Meta- Outcomes* ADVANCE) SR-Liao 2017; IRIS NIDDM) ELIXA, HARMONY CAROLINA (2020), EMPA-Kidney (2022) analysis. -

A Prospective Cohort Study on Effects of Gemigliptin On
www.nature.com/scientificreports OPEN A prospective cohort study on efects of gemigliptin on cardiovascular outcomes in patients with type 2 diabetes (OPTIMUS study) Eun Heui Kim1,9, Sang Soo Kim1,9, Dong Jun Kim2, Young Sik Choi3, Chang Won Lee4, Bon Jeong Ku5, Kwang Soo Cha1, Kee Ho Song6, Dae Kyeong Kim7 & In Joo Kim1,8* This study was performed to evaluate the long-term cardiovascular safety of gemigliptin in patients with type 2 diabetes mellitus (T2DM). After screening, eligible patients with T2DM were enrolled, received gemigliptin, and were followed up for a median of 2.50 years. The primary outcome was a composite of confrmed cardiovascular death, nonfatal myocardial infarction, or nonfatal ischemic stroke (3-point major adverse cardiovascular event [MACE]). The key secondary outcomes were incidence of all-cause mortality and any other cardiovascular events. A total of 5179 patients were included in the study and 5113 were treated with gemigliptin. Overall, the primary outcome occurred in 26 patients within 12 months (estimated incidence by Cox proportional hazard model 0.49%, 95% CI 0.29–0.69%) and in 54 patients within 54 months (estimated incidence from Cox proportional hazard model 1.35%, 95% CI 0.92–1.77%). During the study period, the incidence rates of each component of the primary composite outcome were 0.04% (0.2 events per 1000 person-years) for cardiovascular death, 0.51% (2.2 events per 1000 person-years) for nonfatal myocardial infarction, and 0.61% (2.5 events per 1000 person-years) for nonfatal ischemic stroke. The incidence of all-cause mortality was 0.82% (3.2 events per 1000 person-years) and the incidences of other cardiovascular events were all less than 0.3%. -

Pharmacokinetics and Pharmacodynamics of a Fixed-Dose
Transl Clin Pharmacol. 2020 Mar;28(1):43-54 https://doi.org/10.12793/tcp.2020.28.e2 pISSN 2289-0882·eISSN 2383-5427 Original Article Pharmacokinetics and pharmacodynamics of a fixed-dose combination of gemigliptin/metformin sustained release 25/500 mg compared to the loose combination in healthy male subjects Xuanyou Jin , Eunwoo Kim , Ki Young Huh , Inyoung Hwang , * Received: Feb 20, 2020 Joo-Youn Cho , Kyung-Sang Yu , and SeungHwan Lee Revised: Mar 18, 2020 Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Accepted: Mar 19, 2020 Hospital, Seoul 03080, Korea *Correspondence to SeungHwan Lee Department of Clinical Pharmacology and ABSTRACT Therapeutics, Seoul National University College of Medicine and Hospital, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea. A fixed-dose combination (FDC) of gemigliptin/metformin can improve the medication E-mail: [email protected] adherence in patients with type 2 diabetes mellitus (T2DM). In this study, the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of gemigliptin and metformin were compared Copyright © 2020 Translational and Clinical between FDC and the corresponding loose combination under fasted and fed states. A Pharmacology It is identical to the Creative Commons two-part, randomized, open label, single-dose, two-way crossover study was conducted in Attribution Non-Commercial License (https:// healthy male subjects. Under fasted (part 1) or fed (part 2) state, 2 FDC tablets of gemigliptin/ creativecommons.org/licenses/by-nc/4.0/). metformin sustained release (SR) 25/500 mg or loose combination with one tablet of gemigliptin 50 mg and two tablets of metformin extended release (XR) 500 mg were orally ORCID iDs Xuanyou Jin administered in each period with a 7-day washout.