Regulation of Atp-Sensitive Potassiumchannels in The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Mechanisms Underlying Ketamine-Mediated Inhibition Of
Anesthesiology 2005; 102:93–101 © 2004 American Society of Anesthesiologists, Inc. Lippincott Williams & Wilkins, Inc. Molecular Mechanisms Underlying Ketamine-mediated Inhibition of Sarcolemmal Adenosine Triphosphate- sensitive Potassium Channels Takashi Kawano, M.D.,* Shuzo Oshita, M.D.,† Akira Takahashi, M.D.,‡ Yasuo Tsutsumi, M.D.,* Katsuya Tanaka, M.D.,§ Yoshinobu Tomiyama, M.D., Hiroshi Kitahata, M.D.,# Yutaka Nakaya, M.D.** Background: Ketamine inhibits adenosine triphosphate-sen- sensitive channel pore.3 These channels, as metabolic sitive potassium (KATP) channels, which results in the blocking sensors, are associated with such cellular functions as of ischemic preconditioning in the heart and inhibition of va- insulin secretion, cardiac preconditioning, vasodilata- sorelaxation induced by KATP channel openers. In the current 4–7 Downloaded from http://pubs.asahq.org/anesthesiology/article-pdf/102/1/93/357819/0000542-200501000-00017.pdf by guest on 27 September 2021 study, the authors investigated the molecular mechanisms of tion, and neuroprotection. ketamine’s actions on sarcolemmal KATP channels that are re- In cardiac myocytes, intravenous general anesthetics, associated by expressed subunits, inwardly rectifying potas- such as ketamine racemate, propofol, and thiamylal, di- sium channels (Kir6.1 or Kir6.2) and sulfonylurea receptors 8–10 rectly inhibit native sarcolemmal KATP channels. Al- (SUR1, SUR2A, or SUR2B). though these observations suggest that intravenous an- Methods: The authors used inside-out patch clamp configura- tions to investigate the effects of ketamine on the activities of esthetics may impair the endogenous organ protective reassociated Kir6.0/SUR channels containing wild-type, mutant, mechanisms mediated by KATP channels, the possibility or chimeric SURs expressed in COS-7 cells. -
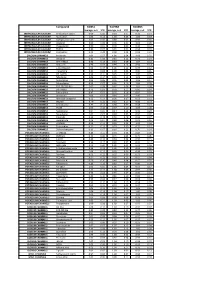
Biomol Average and SD Table S1.Xlsx
Compound GliNS1 G179NS G166NS average, n=5 STD average, n=3 STD average, n=3 STD INTRACELLULAR CALCIUM Antibiotic A-23187 0.00 0.00 0.10 0.07 0.15 0.14 INTRACELLULAR CALCIUM Ryanodine 1.04 0.14 1.03 0.03 1.03 0.03 INTRACELLULAR CALCIUM Cyclopiazonic acid 1.01 0.06 0.88 0.05 0.92 0.06 INTRACELLULAR CALCIUM Gingerol 1.00 0.06 0.91 0.01 1.01 0.06 INTRACELLULAR CALCIUM Thapsigargin 0.00 0.01 0.00 0.00 0.10 0.12 INTRACELLULAR CALCIUM TMB-8 0.89 0.07 0.91 0.05 0.94 0.03 INTRACELLULAR CALCIUM Dantrolene 0.91 0.08 0.98 0.05 0.94 0.01 CALCIUM CHANNELS Amiloride 1.01 0.07 1.01 0.04 1.03 0.05 CALCIUM CHANNELS Benzamil 0.83 0.08 0.83 0.12 0.96 0.04 CALCIUM CHANNELS BAY K-8644 0.93 0.13 0.93 0.09 1.07 0.14 CALCIUM CHANNELS Diltiazem 0.96 0.07 0.99 0.12 0.94 0.14 CALCIUM CHANNELS L-cis-Diltiazem 0.91 0.17 1.01 0.12 0.95 0.12 CALCIUM CHANNELS Flunarizine 0.85 0.08 1.00 0.06 0.85 0.05 CALCIUM CHANNELS FPL-64176 0.99 0.11 0.95 0.07 1.05 0.05 CALCIUM CHANNELS Nifedipine 1.06 0.17 0.95 0.12 1.03 0.09 CALCIUM CHANNELS Nimodipine 1.05 0.06 0.95 0.03 1.06 0.17 CALCIUM CHANNELS Nitrendipine 0.99 0.07 0.96 0.10 1.04 0.09 CALCIUM CHANNELS SDZ-202791 R(-) 1.01 0.08 0.92 0.06 1.01 0.08 CALCIUM CHANNELS SKF-96365 0.73 0.05 0.70 0.11 0.69 0.04 CALCIUM CHANNELS Tetrandrine 0.47 0.07 0.76 0.16 0.87 0.20 CALCIUM CHANNELS Verapamil 1.01 0.02 0.89 0.07 1.06 0.20 CALCIUM CHANNELS Methoxy Verapamil 0.93 0.14 0.96 0.07 0.93 0.13 CALCIUM CHANNELS Bepridil 0.70 0.16 0.92 0.15 0.84 0.14 CALCIUM CHANNELS Amiodarone 0.32 0.12 0.58 0.07 0.48 0.23 CALCIUM CHANNELS YS035 1.00 0.16 -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

Draft Assessment Report on Chelidonium Majus L., Herba
25 November 2010 EMA/HMPC/369801/2009 Committee on Herbal Medicinal Products (HMPC) Assessment report on Chelidonium majus L., herba Based on Article 10a of Directive 2001/83/EC as amended (well-established use) Based on Article 16d(1), Article 16f and Article 16h of Directive 2001/83/EC as amended (traditional use) Draft Herbal substance(s) (binomial scientific name of Chelidonium majus L. the plant, including plant part) Dried whole or cut aerial parts of the plant Herbal preparation(s) Internal Use a) Chelidonii herba: comminuted b) Chelidonii tincture: 1:10 ethanol 45% (V/V) c) Chelidonii extractum fluidum: 1:1 ethanol 25% (V/V) d) Chelidonii extractum siccum (concentration not specified) e) Chelidonium majus mother tincture (M.T. (ø)) External Use a) Eye-drops: (preparation not specified) b) Ointment: (concentration not specified) Pharmaceutical forms Herbal preparation in solid or liquid dosage form or as a herbal tea for oral use. Herbal preparation in solid or liquid dosage form for external use. Rapporteur Assessor(s) 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7523 7051 E-mail [email protected] Website www.ema.europa.eu An agency of the European Union © European Medicines Agency, 2011. Reproduction is authorised provided the source is acknowledged. Note: This Assessment Report is published to support the release for public consultation of the draft Community herbal monograph on Chelidonium majus L. It should be noted that this document is a working document, not yet fully edited, and which shall be further developed after the release for consultation of the monograph. -

Effects of Berberine, Chelerythrine, and Sanguinarine on Proliferation in Four Human Immortalized Cell Lines
Journal of the Iowa Academy of Science: JIAS Volume 119 Number 1-4 Article 6 2012 Effects of Berberine, Chelerythrine, and Sanguinarine on Proliferation in Four Human Immortalized Cell Lines David S. Senchina Drake University Nisarg B. Shah Drake University Marc G. Busch Drake University Let us know how access to this document benefits ouy Copyright © Copyright 2014 by the Iowa Academy of Science, Inc. Follow this and additional works at: https://scholarworks.uni.edu/jias Part of the Anthropology Commons, Life Sciences Commons, Physical Sciences and Mathematics Commons, and the Science and Mathematics Education Commons Recommended Citation Senchina, David S.; Shah, Nisarg B.; and Busch, Marc G. (2012) "Effects of Berberine, Chelerythrine, and Sanguinarine on Proliferation in Four Human Immortalized Cell Lines," Journal of the Iowa Academy of Science: JIAS, 119(1-4), 22-27. Available at: https://scholarworks.uni.edu/jias/vol119/iss1/6 This Research is brought to you for free and open access by the Iowa Academy of Science at UNI ScholarWorks. It has been accepted for inclusion in Journal of the Iowa Academy of Science: JIAS by an authorized editor of UNI ScholarWorks. For more information, please contact [email protected]. Jour. Iowa Acad. Sci. 119(1--4):22-27, 2012 Effects of Berberine, Chelerythrine, and Sanguinarine on Proliferation in Four Human Immortalized Cell Lines DAVIDS. SENCHINA1·*, NISARG B. SHAH2 and MARC G. BUSCH 1 1Deparcmenc of Biology, Drake University, Des Moines, IA 2Pharmacy Program, Drake University, Des Moines, IA Bloodroot (Sanguinaria canadensis L., Papaveraceae) is a plane rich in benzophenanchridine (isoquinoline) alkaloids ~uch_ as sanguinarine and chelerychrine. -

Low Affinity Uniporter Carrier Proteins Can Increase Net Substrate Uptake
www.nature.com/scientificreports OPEN Low afnity uniporter carrier proteins can increase net substrate uptake rate by reducing efux Received: 10 November 2017 Evert Bosdriesz 1,3, Meike T. Wortel 1,4, Jurgen R. Haanstra 1, Marijke J. Wagner1, Accepted: 9 March 2018 Pilar de la Torre Cortés2 & Bas Teusink 1 Published: xx xx xxxx Many organisms have several similar transporters with diferent afnities for the same substrate. Typically, high-afnity transporters are expressed when substrate is scarce and low-afnity ones when it is abundant. The beneft of using low instead of high-afnity transporters remains unclear, especially when additional nutrient sensors are present. Here, we investigate two hypotheses. It was previously hypothesized that there is a trade-of between the afnity and the catalytic efciency of transporters, and we fnd some but no defnitive support for it. Additionally, we propose that for uptake by facilitated difusion, at saturating substrate concentrations, lowering the afnity enhances the net uptake rate by reducing substrate efux. As a consequence, there exists an optimal, external-substrate- concentration dependent transporter afnity. A computational model of Saccharomyces cerevisiae glycolysis shows that using the low afnity HXT3 transporter instead of the high afnity HXT6 enhances the steady-state fux by 36%. We tried to test this hypothesis with yeast strains expressing a single glucose transporter modifed to have either a high or a low afnity. However, due to the intimate link between glucose perception and metabolism, direct experimental proof for this hypothesis remained inconclusive. Still, our theoretical results provide a novel reason for the presence of low-afnity transport systems. -

Label-Free Cell Phenotypic Profiling Decodes the Composition And
OPEN Label-free cell phenotypic profiling SUBJECT AREAS: decodes the composition and signaling POTASSIUM CHANNELS SENSORS AND PROBES of an endogenous ATP-sensitive Received potassium channel 28 January 2014 Haiyan Sun1*, Ying Wei1, Huayun Deng1, Qiaojie Xiong2{, Min Li2, Joydeep Lahiri1 & Ye Fang1 Accepted 24 April 2014 1Biochemical Technologies, Science and Technology Division, Corning Incorporated, Corning, NY 14831, United States of Published America, 2The Solomon H. Snyder Department of Neuroscience and High Throughput Biology Center, Johns Hopkins University 12 May 2014 School of Medicine, Baltimore, Maryland 21205, United States of America. Current technologies for studying ion channels are fundamentally limited because of their inability to Correspondence and functionally link ion channel activity to cellular pathways. Herein, we report the use of label-free cell requests for materials phenotypic profiling to decode the composition and signaling of an endogenous ATP-sensitive potassium should be addressed to ion channel (KATP) in HepG2C3A, a hepatocellular carcinoma cell line. Label-free cell phenotypic agonist Y.F. (fangy2@corning. profiling showed that pinacidil triggered characteristically similar dynamic mass redistribution (DMR) com) signals in A431, A549, HT29 and HepG2C3A, but not in HepG2 cells. Reverse transcriptase PCR, RNAi knockdown, and KATP blocker profiling showed that the pinacidil DMR is due to the activation of SUR2/ Kir6.2 KATP channels in HepG2C3A cells. Kinase inhibition and RNAi knockdown showed that the pinacidil * Current address: activated KATP channels trigger signaling through Rho kinase and Janus kinase-3, and cause actin remodeling. The results are the first demonstration of a label-free methodology to characterize the Biodesign Institute, composition and signaling of an endogenous ATP-sensitive potassium ion channel. -

Consecutive Pharmacological Activation of PKA and PKC Mimics the Potent Cardioprotection of Temperature Preconditioning
Cardiovascular Research (2010) 88, 324–333 doi:10.1093/cvr/cvq190 Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning Igor Khaliulin*, Joanna E. Parker, and Andrew P. Halestrap Department of Biochemistry and the Bristol Heart Institute, University of Bristol, University Walk, Bristol BS8 1TD, UK Received 18 February 2010; revised 14 May 2010; accepted 8 June 2010; online publish-ahead-of-print 16 June 2010 Time for primary review: 25 days Aims Temperature preconditioning (TP) provides very powerful protection against ischaemia/reperfusion. Understanding the signalling pathways involved may enable the development of effective pharmacological cardioprotection. We investigated the interrelationship between activation of protein kinase A (PKA) and protein kinase C (PKC) in the signalling mechanisms of TP and developed a potent pharmacological intervention based on this mechanism. ..................................................................................................................................................................................... Methods Isolated rat hearts were subjected to TP, 30 min global ischaemia, and 60 min reperfusion. Other control and TP and results hearts were perfused with either sotalol (b-adrenergic blocker) or H-89 (PKA inhibitor). Some hearts were pre- treated with either isoproterenol (b-adrenergic agonist) or adenosine (PKC activator) that were given alone, simul- taneously, or sequentially. Pre-treatment with isoproterenol, -

Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level
International Journal of Molecular Sciences Review Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level Sweilem B. Al Rihani 1 , Lucy I. Darakjian 1, Malavika Deodhar 1 , Pamela Dow 1 , Jacques Turgeon 1,2 and Veronique Michaud 1,2,* 1 Tabula Rasa HealthCare, Precision Pharmacotherapy Research and Development Institute, Orlando, FL 32827, USA; [email protected] (S.B.A.R.); [email protected] (L.I.D.); [email protected] (M.D.); [email protected] (P.D.); [email protected] (J.T.) 2 Faculty of Pharmacy, Université de Montréal, Montreal, QC H3C 3J7, Canada * Correspondence: [email protected]; Tel.: +1-856-938-8697 Abstract: The blood–brain barrier (BBB) is a highly selective and restrictive semipermeable network of cells and blood vessel constituents. All components of the neurovascular unit give to the BBB its crucial and protective function, i.e., to regulate homeostasis in the central nervous system (CNS) by removing substances from the endothelial compartment and supplying the brain with nutrients and other endogenous compounds. Many transporters have been identified that play a role in maintaining BBB integrity and homeostasis. As such, the restrictive nature of the BBB provides an obstacle for drug delivery to the CNS. Nevertheless, according to their physicochemical or pharmacological properties, drugs may reach the CNS by passive diffusion or be subjected to putative influx and/or efflux through BBB membrane transporters, allowing or limiting their distribution to the CNS. Drug transporters functionally expressed on various compartments of the BBB involve numerous proteins from either the ATP-binding cassette (ABC) or the solute carrier (SLC) superfamilies. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -
![Arxiv:1912.06275V2 [Q-Bio.BM] 18 Feb 2021](https://docslib.b-cdn.net/cover/6953/arxiv-1912-06275v2-q-bio-bm-18-feb-2021-1116953.webp)
Arxiv:1912.06275V2 [Q-Bio.BM] 18 Feb 2021
General Principles of Secondary Active Transporter Function Oliver Beckstein1, a) and Fiona Naughton1 Department of Physics, Arizona State University, Tempe AZ 85287, USA (Dated: February 19, 2021) Transport of ions and small molecules across the cell membrane against electrochemical gradients is catalyzed by integral membrane proteins that use a source of free energy to drive the energetically uphill flux of the transported substrate. Secondary active transporters couple the spontaneous influx of a “driving” ion such as Na+ or H+ to the flux of the substrate. The thermodynamics of such cyclical non-equilibrium systems are well understood and recent work has focused on the molecular mechanism of secondary active transport. The fact that these transporters change their conformation between an inward-facing and outward-facing conformation in a cyclical fashion, called the alternating access model, is broadly recognized as the molecular framework in which to describe transporter function. However, only with the advent of high resolution crystal structures and detailed computer simulations has it become possible to recognize common molecular-level principles between disparate transporter families. Inverted repeat symmetry in secondary active transporters has shed light on how protein structures can encode a bi-stable two-state system. More detailed analysis (based on experimental structural data and detailed molecular dynamics simulations) indicates that transporters can be understood as gated pores with at least two coupled gates. These gates are not just a convenient cartoon element to illustrate a putative mechanism but map to distinct parts of the transporter protein. Enumerating all distinct gate states naturally includes occluded states in the alternating access picture and also suggests what kind of protein conformations might be observable. -

Plays an Important Role in Drug-Induced Cardiac Arrhythmias: Beyond QT-Prolongation and Torsades De Pointes (Tdps)
Journal of Pharmacological and Toxicological Methods 68 (2013) 250–259 Contents lists available at ScienceDirect Journal of Pharmacological and Toxicological Methods journal homepage: www.elsevier.com/locate/jpharmtox Original article A new biomarker – index of Cardiac Electrophysiological Balance (iCEB) – plays an important role in drug-induced cardiac arrhythmias: beyond QT-prolongation and Torsades de Pointes (TdPs) Hua Rong Lu a,⁎, Gan-Xin Yan b, David J. Gallacher a a Janssen Research and Development, Janssen Pharmaceutica NV, Belgium b Main Line Health Heart Center and Lankenau Institute for Medical Research, Wynnewood, PA, USA article info abstract Article history: Introduction: In the present study, we investigated whether a new biomarker – index of cardiac electro- Received 9 November 2012 physiological balance (iCEB=QT/QRS) – could predict drug-induced cardiac arrhythmias (CAs), including ven- Accepted 5 January 2013 tricular tachycardia/ventricular fibrillation (VT/VF) and Torsades de Pointes (TdPs). Methods: The rabbit left ventricular arterially-perfused-wedge was used to investigate whether the simple iCEB measured from the Keywords: ECG is reflective of the more difficult measurement of λ (effective refractory period×conduction velocity) iCEB (index of Cardiac Electrophysiological for predicting CAs induced by a number of drugs. Results: Dofetilide concentration-dependently increased Balance) iCEB and λ, predicting potential risk of drug-induced incidence of early afterdepolarizations (EADs) starting Drug-induced arrhythmias μ μ μ μ QRS at 0.01 M. Digoxin (1 and 5 M), encainide (5 and 20 M) and propoxyphene (10 and 100 M) markedly re- QT duced both iCEB and λ, predicting their ability to induce non-TdP-like VT/VF.