Complimentary and Personal Copy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Distillation 65 Chem 355 Jasperse DISTILLATION
Distillation 65 Chem 355 Jasperse DISTILLATION Background Distillation is a widely used technique for purifying liquids. The basic distillation process involves heating a liquid such that liquid molecules vaporize. The vapors produced are subsequently passed through a water-cooled condenser. Upon cooling, the vapor returns to it’s liquid phase. The liquid can then be collected. The ability to separate mixtures of liquids depends on differences in volatility (the ability to vaporize). For separation to occur, the vapor that is condensed and collected must be more pure than the original liquid mix. Distillation can be used to remove a volatile solvent from a nonvolatile product; to separate a volatile product from nonvolatile impurities; or to separate two or more volatile products that have sufficiently different boiling points. Vaporization and Boiling When a liquid is placed in a closed container, some of the molecules evaporate into any unoccupied space in the container. Evaporation, which occurs at temperatures below the boiling point of a compound, involves the transition from liquid to vapor of only those molecules at the liquid surface. Evaporation continues until an equilibrium is reached between molecules entering and leaving the liquid and vapor states. The pressure exerted by these gaseous molecules on the walls of the container is the equilibrium vapor pressure. The magnitude of this vapor pressure depends on the physical characteristics of the compound and increases as temperature increases. In an open container, equilibrium is never established, the vapor can simply leave, and the liquid eventually disappears. But whether in an open or closed situation, evaporation occurs only from the surface of the liquid. -

Experiment 2 — Distillation and Gas Chromatography
Chem 21 Fall 2009 Experiment 2 — Distillation and Gas Chromatography _____________________________________________________________________________ Pre-lab preparation (1) Read the supplemental material on distillation theory and techniques from Zubrick, The Organic Chem Lab Survival Manual, and the section on Gas Chromatography from Fessenden, Fessenden, and Feist, Organic Laboratory Techniques, then read this handout carefully. (2) In your notebook, write a short paragraph summarizing what you will be doing in this experiment and what you hope to learn about the efficiencies of the distillation techniques. (3) Sketch the apparatus for the simple and fractional distillations. Your set-up will look much like that shown on p 198 of Zubrick, except that yours will have a simple drip tip in place of the more standard vacuum adaptor. (4) Look up the structures and relevant physical data for the two compounds you will be using. What data are relevant? Read the procedure, think about the data analysis, and decide what you need. (5) Since you have the necessary data, calculate the log of the volatility factor (log α) that you will need for the theoretical plate calculation. Distillation has been used since antiquity to separate the components of mixtures. In one form or another, distillation is used in the manufacture of perfumes, flavorings, liquors, and a variety of other organic chemicals. One of its most important modern applications is in refining crude oil to make fuels, lubricants, and other petrochemicals. The first step in the refining process is separation of crude petroleum into various hydrocarbon fractions by distillation through huge fractionating columns, called distillation towers, that are hundreds of feet high. -

Distillation1
Distillation1 Distillation is a commonly used method for purifying liquids and separating mixtures of liquids into their individual components. Familiar examples include the distillation of crude fermentation broths into alcoholic spirits such as gin and vodka, and the fractionation of crude oil into useful products such as gasoline and heating oil. In the organic lab, distillation is used for purifying solvents and liquid reaction products. In analyzing a distillation, how do we know the real composition of each collected component? In this lab, we will introduce gas chromatography (GC), which will tell us how pure each fraction we collected is. After the distillation of your unknown is complete, you will analyze both components via GC. See page 11 and 12 for a light discussion on GC. To understand distillation, first consider what happens upon heating a liquid. At any temperature, some molecules of a liquid possess enough kinetic energy to escape into the vapor phase (evaporation) and some of the molecules in the vapor phase return to the liquid (condensation). An equilibrium is set up, with molecules going back and forth between liquid and vapor. At higher temperatures, more molecules possess enough kinetic energy to escape, which results in a greater number of molecules being present in the vapor phase. If the liquid is placed into a closed container with a pressure gauge attached, one can obtain a quantitative measure of the degree of vaporization. This pressure is defined as the vapor pressure of the compound, which can be measured at different temperatures. Consider heating cyclohexane, a liquid hydrocarbon, and measuring its vapor pressure at different temperatures. -

A Large Spinning-Band Fractionating Column for Use with Small Quantities of Liquids
468 INDUSTRIAL AND ENGINEERING CHEMISTRY VOL. 12, h-0.8 Operation of Viscometer After a viscosity measurement has been made, the liquid The filling operation is effected by means of the filling tube may be removed readily by applying a vacuum to the leveling illustrated in Figure 3. The asphaltic liquid is warmed to a tube. Benzene or carbon tetrachloride may be introduced temperature (60' to 82.22' C., 140" to 180OF.) sufficient to into the left arm, and as it is sucked through the instrument it ymit its being poured into the 100-mesh copper funnel sieve. he liquid passes through the sieve into the receiver arm of the sweeps the viscometer clean of oil. As the vacuum is con- viscometer and catches at point P where the narrow connecting tinued, the cleaning solvent is soon evaporated. The cleaning tube joins the receiver tube reservoir. The flow down through operation is thus effected simply without removal of the the connecting tubing is controlled by means of pressure changes viscometer from the thermostat bath. caused by a roller device (devised by V. Lantz, of these labora- tories, and shown in Figure 2) which squeezes a rubber tubing The viscometer is readily calibrated by the use of oils of connected to the left arm of the viscometer. To control the rate known viscosity (such as the alpha and beta oils of the Ameri- of flow from the filling tube into the viscometer and to adjust can Petroleum Institute) at low enough temperatures to get the flow so that the upper surface of the liquid approaches mark good flow times. -

11.4. the Fractionating Column
DISTILLATION 559 vapour to be obtained that is substantially richer in the more volatile component than is the liquid left in the still. This is achieved by an arrangement known as a fractionating column which enables successive vaporisation and condensation to be accomplished in one unit. Detailed consideration of this process is given in Section 11.4. 11.3.4. Batch distillation In batch distillation, which is considered in detail in Section 11.6, the more volatile component is evaporated from the still which therefore becomes progressively richer in the less volatile constituent. Distillation is continued, either until the residue of the still contains a material with an acceptably low content of the volatile material, or until the distillate is no longer sufficiently pure in respect of the volatile content. 11.4. THE FRACTIONATING COLUMN 11.4.1. The fractionating process The operation of a typical fractionating column may be followed by reference to Figure 11.11. The column consists of a cylindrical structure divided into sections by Figure 11.11. Continuous fractionating column with rectifying and stripping sections 560 CHEMICAL ENGINEERING a series of perforated trays which permit the upward flow of vapour. The liquid reflux flows across each tray, over a weir and down a downcomer to the tray below. The vapour rising from the top tray passes to a condenser and then through an accumulator or reflux drum and a reflux divider, where part is withdrawn as the overhead product D, and the remainder is returned to the top tray as reflux R. The liquid in the base of the column is frequently heated, either by condensing steam or by a hot oil stream, and the vapour rises through the perforations to the bottom tray. -

Chem 2219: Exp. #2 Fractional Distillation
Chem 2219: Exp. #2 Fractional Distillation Objective: In this experiment you will learn to separate the components of a solvent mixture (two liquids) by distillation. The boiling point (BP) range and refractive index (RI) will be determined for the three fractions. Gas chromatography will be used to determine the percent composition and purity of the first fraction. * * Gas Chromatography is extremely useful in determining the percent composition/purity of known liquids. Recall the boiling point and refractive index (R.I.) are two useful physical properties of a liquid. They are used for identification purposes. The R.I. is additionally used as a measure of purity of the sample being examined. Reading Assignment: MTOL, pp. 79-83 (distillation theory), 57-62 (gas chromatography) and: OCLT, pp. 254-262 (separation theory), 279-281 (fractional distillation), 139-153 (gas chromatography). Also on CANVAS read and answer prelab questions for – Fractional Distillation and (prelab for next week) Gas Chromatography Concepts: Boiling Point, Condensate, Condensation, Distillate, Distillation, Evaporation, Reflux, Refractive Index, Theoretical Plates, Vaporization Chemicals: Cyclohexane, Toluene, p-Xylene Safety Precautions: Wear chemical splash-proof goggles and appropriate attire at all times. Cyclohexane, toluene and p-xylene are flammable liquids. Hot glassware looks just like cold glassware. Be careful when working with hot glassware! Do not to touch it! Materials: aluminum block, aluminum foil, beaker (100ml), Claisen head adapter, conical vial (5 ml), disposable pipets & bulbs, disposable vials and lids, finger clamps (2), glass wool batting, Hickman still head, hot plate with magnetic stirrer, labels (3-small), magnetic spin vane (large), ringstand, Teflon septum with hole in the center, and thermometer Instruments: Refractometer, Gas Chromatograph (maybe), Magritek desktop NMR (cbolon updated 201030) 1 Chem 2219: Exp. -

Distillation Columns - Fractioning Columns
Distillation Columns - Fractioning Columns Distillation is the process of separating two or more miscible liquids by taking advantage of the boiling point differences between the liquids. For methanol and water, heat is added to the mixture of methanol and water and eventually the most volatile component (methanol) begins to vaporize. As the methanol vaporizes it takes with it molecules of water. The methanol-water vapor mixture is then condensed and evaporated again, giving a higher mole fraction of methanol in the vapor phase and a higher mole fraction of water in the liquid phase. This process of condensation and evaporation continues in stages up the column until the methanol rich vapor component is condensed and collected as tops product (99.5% recovery / 99.5% pure) and the IPA/Water rich liquid is collected as bottoms product. What is Fractionating columns SRS Engineering Corporation is premier manufacturer of distillation columns for various applications. Fractional distillation is a method used to separate liquids with similar boiling points. This method relies upon a gradient of temperatures existing in the condenser stage of the equipment. A vertical condenser is often used in the fractional distillation technique. Extracting products in the liquid stage at the different heights of the column makes it possible for the extraction of liquids that have different boiling points. The greater the distance over which the temperature gradient in the condenser is applied, the easier and the greater separation is. Heating under Reflex Heating under reflex allows the mixture including volatile materials to be heated for a long time without loosing any solvent. -
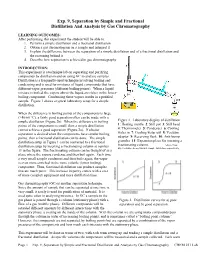
Exp. 9, Separation by Simple and Fractional Distillation and Analysis by Gas Chromatography
Exp. 9, Separation by Simple and Fractional Distillation And Analysis by Gas Chromatography LEARNING OUTCOMES: After performing this experiment the student will be able to: 1. Perform a simple distillation and a fractional distillation 2. Obtain a gas chromatogram on a sample and interpret it 3. Explain the difference between the separation of a simple distillation and of a fractional distillation and the reasoning behind it 4. Describe how separation is achieved in gas chromatography INTRODUCTION: This experiment is a technique lab on separating and purifying compounds by distillation and on using GC to analyze samples. Distillation is a frequently-used technique involving boiling and condensing and is used for mixtures of liquid compounds that have different vapor pressures (different boiling points). When a liquid mixture is boiled, the vapors above the liquid are richer in the lower- boiling component. Condensing these vapors results in a purified sample. Figure 1 shows a typical laboratory setup for a simple distillation. When the difference in boiling points of the components is large (>40-60 °C), a fairly good separation often can be made with a simple distillation (Figure 2b). When the difference in boiling Figure 1. Laboratory display of distillation: points of the components is small, then a simple distillation 1: Heating mantle 2: Still pot 3: Still head cannot achieve a good separation (Figure 2a). If a better 4: Thermometer 5: Condenser 6: Cooling separation is desired when the components have similar boiling water in 7: Cooling water out 8: Vacuum points, then a fractional distillation can be done. The simple adaptor 9: Receiving flask 10: Anti-bump distillation setup in Figure 1 can be converted to a fractional granules 11: Expansion place for inserting a distillation setup by inserting a fractionating column at number fractionating column. -

Chemistry Continued…… (7) Fractional Distillation It Is
Class: IX Topic: Is Matter Around Us Pure Subject: Chemistry Date -14/07/2020 Continued…… (7) Fractional distillation It is the process of separating two or more miscible liquids by distillation, the distillate being collected in fractions due to boiling at different temperatures. Fractionating Column: The apparatus used in this process is similar to that for simple distillation except a fractionating column which is fitted in between the distillation flask and the condenser. A simple fractionating colunrn is a tube packed with glass beads. The beads provide surface for the vapours to cool and condense repeatedly. Principle of Fractional Distillation: In a mixture of two or more miscible liquids, the separation of various liquids depends on their boilibg points. The liquid having lower boiling point boils first and can be obtained first from the fractionating column than the liquid having higher boiling point. Applications of Fractional Distillation: • It is used to separate a mixture of miscible liquids like alcohol-water mixture. • It is used to separate cruid oil ‘petroleum’ into useful fractions like kerosene, petrol, diesel, etc. • It is used to separate different gases of the air by taking the liquid air. • • Separation of components of air (8) Crystallisation : Crystallisation is a process used to separates a pure solid in the form of its crystals from a solution. The process involves cooling a hot, concentrated solution of a substance to obtain crystals. Applications of Crystallisation: • Purification of common salt obtained from sea water. • To obtain crystals of alum (phitkari) from impure samples. • To obtain pure copper sulphate from an impure sample. -

Module 5: Distillation
NPTEL – Chemical – Mass Transfer Operation 1 MODULE 5: DISTILLATION LECTURE NO. 1 5.1. Introduction Distillation is method of separation of components from a liquid mixture which depends on the differences in boiling points of the individual components and the distributions of the components between a liquid and gas phase in the mixture. The liquid mixture may have different boiling point characteristics depending on the concentrations of the components present in it. Therefore, distillation processes depends on the vapor pressure characteristics of liquid mixtures. The vapor pressure is created by supplying heat as separating agent. In the distillation, the new phases differ from the original by their heat content. During most of the century, distillation was by far the most widely used method for separating liquid mixtures of chemical components (Seader and Henley, 1998). This is a very energy intensive technique, especially when the relative volatility of the components is low. It is mostly carried out in multi tray columns. Packed column with efficient structured packing has also led to increased use in distillation. 5.1.1. Vapor Pressure Vapor Pressure: The vaporization process changes liquid to gaseous state. The opposite process of this vaporization is called condensation. At equilibrium, the rates of these two processes are same. The pressure exerted by the vapor at this equilibrium state is termed as the vapor pressure of the liquid. It depends on the temperature and the quantity of the liquid and vapor. From the following Clausius-Clapeyron Equation or by using Antoine Equation, the vapor pressure can be calculated. Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 9 NPTEL – Chemical – Mass Transfer Operation 1 Clausius-Clapeyron Equation: pv 1 1 ln (5.1) v p1 R T1 T v v where p and p1 are the vapor pressures in Pascal at absolute temperature T and T1 in K. -

United States Patent (19) 11 Patent Number: 4,770,746 Mayo Et Al
United States Patent (19) 11 Patent Number: 4,770,746 Mayo et al. (45) Date of Patent: Sep. 13, 1988 (54). SPINNING BAND FRACTIONATING 2,712,520 7/1955 Nester ........................... 203/DIG. 2 COLUMN 2,764,534 9/1956 Nerheinn .............................. 202/153 2,783,401 2/1957 Foster et al. ..... ... 202/53 75) Inventors: Dana W. Mayo, Brunswick, Me.; 3,002,897 10/1961 Kirkland et al. ... 202/190 Ronald M. Pike, Pelham; Robert J. 3,080,303 3/1963 Nerheim .............................. 202/161 Hinkle, Hampton, both of N.H. OTHER PUBLICATIONS 73 Assignee: Microscale Organic Laboratory Corporation, New Castle, N.H. Perkin-Elmer, Model 131T, "A Compact, Complete 30-Plate Teflon Spinning Band Distillation Unit, Mayo 21 Appl. No.: 518 et al., Microscale Organic Laboratory', John Wileys & (22 Filed: Jan. 5, 1987 Sons, 1986, p. 46. Stinson, Stephen, "New Instrumental Techniques 51) Int. Cl." ............................................... B01D 3/10 Debut at Pittsburgh Conference", Chem. Engineering 52 U.S. C. .................................... 202/153; 202/161; 202/205; 202/237; 202/238; 202/267.1; 203/86; News, Mar. 30, 1987, pp. 20 and 21. 203/91; 203/DIG. 2; 159/DIG. 7; 159/DIG. Primary Examiner-David L. Lacey 16 Assistant Examiner-V. Manoharan 58 Field of Search ................... 202/161, 153,267 R, Attorney, Agent, or Firm-Thomas N. Tarrant 202/205, 237,238, 189, 185.1; 203/DIG. 2, 86, (57) ABSTRACT -- 91, 98; 159/DIG. 7, DIG. 16 Spinning band fractionating column apparatus is dis (56) References Cited closed having a spinning band formed with a magnet U.S. PATENT DOCUMENTS embedded in its bottom end in the manner of a magnetic 2,251,185 7/1941 Carter et al. -

The Carriage Still an Advanced Fractionating Still
The Carriage Still Published in Canada in September 2003 by Saguenay International 17 Hudson Club Rd Rigaud, QC Canada J0P 1P0 Second Edition Copyright © September 2003 by John Stone All rights reserved. No part of this publication, printed or electronic, may be reproduced or transmitted to a third party in any form or by any means, including electronic, without the prior written permission of the author. ISBN 0-9682280-4-6 Contact: John Stone 17 Hudson Club Rd Rigaud, QC Canada J0P 1P0 Telephone: (450) 451-0644 Fax: (450) 451-7699 e-mail: [email protected] Photographs and illustrations by Jim Low, Hudson, QC i Foreword After 17 years of designing small reflux stills for use by amateurs in the home, making major changes here, cosmetic changes there, sometimes doing nothing more than making a change for the sake of change, we feel that its time to freeze the design and make it available immediately. Amateur distillers around the world can then start to build one for themselves and enjoy the fruits of their labour. The Carriage Still combines all the best features of the many designs developed over the years and should satisfy the needs of the most fastidious connoisseurs of high quality spirits. ii TABLE OF CONTENTS Page No. Introduction 1 Types of still The alembic 3 Laboratory pot still 3 Laboratory fractionating still (Vigreux) 4 Amateur glass fractionating still (Stone) 6 The Principles of Distillation 7 Simple distillation --- the pot still 8 Fractional distillation using reflux 8 Health & Safety Poisoning oneself 12 Headaches & hangovers 13 Fire & explosions 14 The Carriage Still The evolution of the still 15 The boiler 17 Power supply 18 The column 18 The stream-splitter 19 Still-head 20 Cooling coil 21 The packing 23 Water supply 24 Column support 25 Collection of alcohol 25 Procedures Production of pure alcohol (vodka) 26 Stage 1 --- Beer stripping 28 Stage 2 --- Rectification.