SYNTHESIS Template V2.0
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

European Patent Office of Opposition to That Patent, in Accordance with the Implementing Regulations
(19) TZZ _T (11) EP 2 686 288 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07C 217/56 (2006.01) C07C 217/62 (2006.01) 25.03.2015 Bulletin 2015/13 C07C 227/36 (2006.01) C07C 57/15 (2006.01) C07C 213/08 (2006.01) C07C 219/28 (2006.01) (2006.01) (21) Application number: 11796817.2 C07B 57/00 (22) Date of filing: 11.11.2011 (86) International application number: PCT/IB2011/055039 (87) International publication number: WO 2012/137047 (11.10.2012 Gazette 2012/41) (54) A PROCESS FOR PREPARING FESOTERODINE PROZESS ZUR HERSTELLUNG VON FESOTERODIN PROCÉDÉ DE PRÉPARATION DE FÉSOTÉRODINE (84) Designated Contracting States: • PATIL, Chetan AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Gujarat GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO Vadodara 390003 (IN) PL PT RO RS SE SI SK SM TR • PATEL, Ronak Gujarat (30) Priority: 07.04.2011 IN MM11722011 Vadodara 390003 (IN) • RAVAL, Prashant (43) Date of publication of application: Gujarat 22.01.2014 Bulletin 2014/04 Vadodara 390003 (IN) • PAREKH, Viral (73) Proprietor: Alembic Pharmaceuticals Limited Gujarat Vadodara 390003 (IN) Vadodara 390003 (IN) • SHAH, Hiral (72) Inventors: Gujarat • RAMAN, Jayaraman, Venkat Vadodara 390003 (IN) Gujarat Vadodara 390003 (IN) (74) Representative: Watson, Robert James et al • PATEL, Samir Mewburn Ellis LLP Gujarat 33 Gutter Lane Vadodara 390003 (IN) London EC2V 8AS (GB) • THAKOR, Indrajit Gujarat (56) References cited: Vadodara 390003 (IN) EP-A1- 2 281 801 WO-A2-2009/126844 • LADANI, Mahesh Gujarat Vadodara 390003 (IN) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Distillation 65 Chem 355 Jasperse DISTILLATION
Distillation 65 Chem 355 Jasperse DISTILLATION Background Distillation is a widely used technique for purifying liquids. The basic distillation process involves heating a liquid such that liquid molecules vaporize. The vapors produced are subsequently passed through a water-cooled condenser. Upon cooling, the vapor returns to it’s liquid phase. The liquid can then be collected. The ability to separate mixtures of liquids depends on differences in volatility (the ability to vaporize). For separation to occur, the vapor that is condensed and collected must be more pure than the original liquid mix. Distillation can be used to remove a volatile solvent from a nonvolatile product; to separate a volatile product from nonvolatile impurities; or to separate two or more volatile products that have sufficiently different boiling points. Vaporization and Boiling When a liquid is placed in a closed container, some of the molecules evaporate into any unoccupied space in the container. Evaporation, which occurs at temperatures below the boiling point of a compound, involves the transition from liquid to vapor of only those molecules at the liquid surface. Evaporation continues until an equilibrium is reached between molecules entering and leaving the liquid and vapor states. The pressure exerted by these gaseous molecules on the walls of the container is the equilibrium vapor pressure. The magnitude of this vapor pressure depends on the physical characteristics of the compound and increases as temperature increases. In an open container, equilibrium is never established, the vapor can simply leave, and the liquid eventually disappears. But whether in an open or closed situation, evaporation occurs only from the surface of the liquid. -

UCLA Electronic Theses and Dissertations
UCLA UCLA Electronic Theses and Dissertations Title Novel microfluidic technologies for the concentration of radionuclides and radiotracers for positron emission tomography Permalink https://escholarship.org/uc/item/7qb3p6x1 Author Chao, Philip Hong-Sean Publication Date 2019 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Los Angeles Novel microfluidic technologies for the concentration of radionuclides and radiotracers for positron emission tomography A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Philip Hong-Sean Chao 2019 © Copyright by Philp Hong-Sean Chao 2019 ABSTRACT OF THE DISSERTATION Novel microfluidic technologies for the concentration of radionuclides and radiotracers for positron emission tomography by Philip Hong-Sean Chao Doctor of Philosophy in Bioengineering University of California, Los Angeles, 2019 Professor Pei-Yu Chiou, Co-Chair Professor Robert Michael van Dam, Co-Chair Positon emission tomography (PET) is an imaging modality capable of visualizing biomolecules in vivo and can be used to aid in disease diagnosis, staging of disease severity, and monitoring of disease response to treatment. PET relies on the use of tracers (i.e. biomolecules labeled with radionuclides) for imaging. Due to the short half-life of the radionuclides used, PET tracer production is typically performed right before an imaging event. Production of a PET tracer can be broken down into three major parts: production of the radionuclide, radiochemical synthesis of the tracer, and, lastly, purification, formulation and quality control testing of the tracer. Several groups, including our own, have looked into leveraging the benefits of microfluidics (reduced system size, finer control of reaction parameters, reduced reagent consumption) towards the production of PET tracers. -

Experiment 2 — Distillation and Gas Chromatography
Chem 21 Fall 2009 Experiment 2 — Distillation and Gas Chromatography _____________________________________________________________________________ Pre-lab preparation (1) Read the supplemental material on distillation theory and techniques from Zubrick, The Organic Chem Lab Survival Manual, and the section on Gas Chromatography from Fessenden, Fessenden, and Feist, Organic Laboratory Techniques, then read this handout carefully. (2) In your notebook, write a short paragraph summarizing what you will be doing in this experiment and what you hope to learn about the efficiencies of the distillation techniques. (3) Sketch the apparatus for the simple and fractional distillations. Your set-up will look much like that shown on p 198 of Zubrick, except that yours will have a simple drip tip in place of the more standard vacuum adaptor. (4) Look up the structures and relevant physical data for the two compounds you will be using. What data are relevant? Read the procedure, think about the data analysis, and decide what you need. (5) Since you have the necessary data, calculate the log of the volatility factor (log α) that you will need for the theoretical plate calculation. Distillation has been used since antiquity to separate the components of mixtures. In one form or another, distillation is used in the manufacture of perfumes, flavorings, liquors, and a variety of other organic chemicals. One of its most important modern applications is in refining crude oil to make fuels, lubricants, and other petrochemicals. The first step in the refining process is separation of crude petroleum into various hydrocarbon fractions by distillation through huge fractionating columns, called distillation towers, that are hundreds of feet high. -

Tailoring Diesel Bioblendstock from Integrated Catalytic Upgrading of Carboxylic Acids: a “Fuel Property First” Approach Xiangchen Huoa,B, Nabila A
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is © The Royal Society of Chemistry 2019 Tailoring Diesel Bioblendstock from Integrated Catalytic Upgrading of Carboxylic Acids: A “Fuel Property First” Approach Xiangchen Huoa,b, Nabila A. Huqa, Jim Stunkela, Nicholas S. Clevelanda, Anne K. Staracea, Amy E. Settlea,b, Allyson M. Yorka,b, Robert S. Nelsona, David G. Brandnera, Lisa Foutsa, Peter C. St. Johna, Earl D. Christensena, Jon Lueckea, J. Hunter Mackc, Charles S. McEnallyd, Patrick A. Cherryd, Lisa D. Pfefferled, Timothy J. Strathmannb, Davinia Salvachúaa, Seonah Kima, Robert L. McCormicka, Gregg T. Beckhama, Derek R. Vardona* aNational Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO, United States bColorado School of Mines, 1500 Illinois St., Golden, CO, United States cUniversity of Massachusetts Lowell, 220 Pawtucket St., Lowell, MA, United States dYale University, 9 Hillhouse Avenue, New Haven, CT, United States *[email protected] Section S1: Experimental Methods Catalyst synthesis and characterization Pt/Al2O3 catalyst was prepared by strong electrostatic adsorption method with chloroplatinic acid hexahydrate as Pt precursor. Al2O3 of 30-50 mesh and Pt precursor were added to deionized water, and solution pH was adjusted to 3 by adding HCl. After stirring overnight, the catalyst particles were recovered by filtration extensively washed with -1 deionized water. The catalyst was dried in the air and reduced in flowing H2 (200 mL min ) at 300°C for 4 h. BET surface area was determined by nitrogen physisorption using a Quadrasorb SI™ surface area analyzer from Quantachrome Instruments. Samples of ~80–120 mg were measured using a 55-point nitrogen adsorption/desorption curve at -196°C. -

Rotary Evaporator (Single/Dual Condenser Type)
Version : 2020 Rotary Evaporator (Single/Dual Condenser Type) USA-RE20-UL/USA-RE20D-UL USA-RE50-UL/USA-RE50D-UL Introduction Rotary evaporator produced by USA LAB Inc. is a new type of product developed by the company for many years of design and production experience. The laboratory instrument rotary evaporator is mainly used to continuously distill a large amount of volatile solvents under reduced pressure. In particular, the concentration of the extract and the distillation of the receiving liquid at the time of chromatographic separation can be used to purify the reaction product. The basic principle of a rotary evaporator is vacuum distillation, that is, under reduced pressure, the distillation flask is continuously rotated as the solvent is distilled. The company's after-sales service will accompany you from the date of purchase. Please refer to this instruction manual (operation manual) before using. If you have any questions, please feel free to contact us and we will do our best to serve you. TABLE OF CONTENTS 1. Working principle and scope of application 4 2. Precautions 4 3. Structure and composition 7 3.1 Outside view 7 3.2 Operating Instructions for operating panel (Figure 6) 12 3.3 Operation instructions 13 3.4 Operation of over-temperature protector 14 4. Installation 15 4.1 Stripping 15 4.2 Installation diagram of upright rod 15 4.3 Installation diagram of three-way flask, rotating shaft, 16 evaporating flask 4.4 Installation diagram of main condenser, receiving 17 flask and check valve Other parts installation 19 5. Specification 20 6. -

Alembic Pot Still
ALEMBIC POT STILL INSTRUCTION MANUAL CAN BE USED WITH THE GRAINFATHER OR T500 BOILER SAFETY Warning: This system produces a highly flammable liquid. PRECAUTION: • Always use the Alembic Pot Still System in a room with adequate ventilation. • Never leave the Alembic Pot Still system unattended when operating. • Keep the Alembic Pot Still system away from all sources of ignition, including smoking, sparks, heat, and open flames. • Ensure all other equipment near to the Alembic Pot Still system or the alcohol is earthed. • A fire extinguishing media suitable for alcohol should be kept nearby. This can be water fog, fine water spray, foam, dry powder, carbon dioxide, sand or dolomite. • Do not boil dry. In the event the still is boiled dry, reset the cutout button under the base of the still. In the very unlikely event this cutout fails, a fusible link gives an added protection. IN CASE OF SPILLAGE: • Shut off all possible sources of ignition. • Clean up spills immediately using cloth, paper towels or other absorbent materials such as soil, sand or other inert material. • Collect, seal and dispose accordingly • Mop area with excess water. CONTENTS Important points before getting started ............................................................................... 3 Preparing the Alembic Pot Still ................................................................................................. 5 Distilling a Whiskey, Rum or Brandy .......................................................................................7 Distilling neutral -

Minimum Reflux in Liquid–Liquid Extraction
17th European Symposium on Computer Aided Process Engineering – ESCAPE17 V. Plesu and P.S. Agachi (Editors) © 2007 Elsevier B.V. All rights reserved. 1 Minimum Reflux in Liquid–Liquid Extraction Santanu Bandyopadhyaya and Calin-Cristian Cormosb aEnergy Systems Engineering, Indian Institute of Technology, Bombay, Powai, Mumbai 400 076, India, E-mail: [email protected] bDepartment of Chemical Engineering, Faculty of Chemistry and Chemical Engineering, University "Babeş-Bolyai", Arany Janos 11, Cluj-Napoca 400028, Romania, E-mail: [email protected] Abstract In a simple countercurrent arrangement of different stages of liquid–liquid extraction operation, the richest extract leaving the operation is in equilibrium with the feed. However, by using reflux it is possible to enrich the extract further. A simple counter current liquid–liquid extraction operation with reflux is analogous in its essentials to distillation. In this paper, the method based on Invariant Rectifying-Stripping (IRS) curves, originally proposed to calculate minimum reflux and minimum energy requirement in distillation, has been extended to liquid–liquid extraction. The equivalent IRS curves for a ternary liquid–liquid extraction predicts the feed location in the counter current process. It also predicts the minimum reflux requirement for a given separation and minimum amount of solvent required. Keywords: liquid extraction, IRS curves, feed location, minimum solvent. 1. Introduction Liquid–liquid extraction is a separation process that takes advantage of the distribution of a substance between two insoluble liquids phases [1]. Feed to be separated is mixed with extracting solvent to produce a solvent-rich phase, called extract, and a solvent-lean phase, called raffinate. -

Reflux Condensation in Narrow Rectangular Channels with Perforated Fins Nadia Souidi, André Bontemps
Reflux condensation in narrow rectangular channels with perforated fins Nadia Souidi, André Bontemps To cite this version: Nadia Souidi, André Bontemps. Reflux condensation in narrow rectangular channels with perforated fins. Applied Thermal Engineering, Elsevier, 2003, 23, pp.871-891. 10.1016/S1359-4311(03)00021-8. hal-00184135 HAL Id: hal-00184135 https://hal.archives-ouvertes.fr/hal-00184135 Submitted on 19 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Reflux condensation in narrow rectangular channels with perforated fins N. Souidi a, A. Bontemps b,* a GRETh-CEA Grenoble, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France b LEGI-GRETh, Universitee Joseph Fourier, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France Reflux condensation is an industrial process that aims to reduce the content of the less volatile com- ponent or to eliminate the non-condensable phase of a vapour mixture, by the means of separation. Separation consists in condensing the less volatile phase and to recover the condensate while simulta- neously, the non-condensable species are recuperated at the top of the system. Compact plate-fin heat exchangers can be used in gas separation processes. -
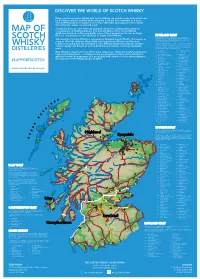
2019 Scotch Whisky
©2019 scotch whisky association DISCOVER THE WORLD OF SCOTCH WHISKY Many countries produce whisky, but Scotch Whisky can only be made in Scotland and by definition must be distilled and matured in Scotland for a minimum of 3 years. Scotch Whisky has been made for more than 500 years and uses just a few natural raw materials - water, cereals and yeast. Scotland is home to over 130 malt and grain distilleries, making it the greatest MAP OF concentration of whisky producers in the world. Many of the Scotch Whisky distilleries featured on this map bottle some of their production for sale as Single Malt (i.e. the product of one distillery) or Single Grain Whisky. HIGHLAND MALT The Highland region is geographically the largest Scotch Whisky SCOTCH producing region. The rugged landscape, changeable climate and, in The majority of Scotch Whisky is consumed as Blended Scotch Whisky. This means as some cases, coastal locations are reflected in the character of its many as 60 of the different Single Malt and Single Grain Whiskies are blended whiskies, which embrace wide variations. As a group, Highland whiskies are rounded, robust and dry in character together, ensuring that the individual Scotch Whiskies harmonise with one another with a hint of smokiness/peatiness. Those near the sea carry a salty WHISKY and the quality and flavour of each individual blend remains consistent down the tang; in the far north the whiskies are notably heathery and slightly spicy in character; while in the more sheltered east and middle of the DISTILLERIES years. region, the whiskies have a more fruity character. -

United States Patent (10) Patent No.: US 9,586,922 B2 Wood Et Al
USOO9586922B2 (12) United States Patent (10) Patent No.: US 9,586,922 B2 Wood et al. (45) Date of Patent: Mar. 7, 2017 (54) METHODS FOR PURIFYING 9,388,150 B2 * 7/2016 Kim ..................... CO7D 307/48 5-(HALOMETHYL)FURFURAL 9,388,151 B2 7/2016 Browning et al. 2007/O161795 A1 7/2007 Cvak et al. 2009, 0234142 A1 9, 2009 Mascal (71) Applicant: MICROMIDAS, INC., West 2010, 0083565 A1 4, 2010 Gruter Sacramento, CA (US) 2010/0210745 A1 8/2010 McDaniel et al. 2011 O144359 A1 6, 2011 Heide et al. (72) Inventors: Alex B. Wood, Sacramento, CA (US); 2014/01 00378 A1 4/2014 Masuno et al. Shawn M. Browning, Sacramento, CA 2014/O1878O2 A1 7/2014 Mikochik et al. (US);US): Makoto N. Masuno,M. s Elk Grove, s 2015/02668432015,0203462 A1 9/20157/2015 CahanaBrowning et etal. al. CA (US); Ryan L. Smith, Sacramento, 2016/0002190 A1 1/2016 Browning et al. CA (US); John Bissell, II, Sacramento, 2016,01681 07 A1 6/2016 Masuno et al. CA (US) 2016/02O7897 A1 7, 2016 Wood et al. (73) Assignee: MICROMIDAS, INC., West FOREIGN PATENT DOCUMENTS Sacramento, CA (US) CN 101475544. A T 2009 (*) Notice: Subject to any disclaimer, the term of this N 1939: A 658 patent is extended or adjusted under 35 DE 635,783 C 9, 1936 U.S.C. 154(b) by 0 days. EP 291494 A2 11, 1988 EP 1049657 B1 3, 2003 (21) Appl. No.: 14/852,306 GB 1220851 A 1, 1971 GB 1448489 A 9, 1976 RU 2429234 C2 9, 2011 (22) Filed: Sep. -

Hydro-Distillation and Steam Distillation from Aromatic Plants
Hydro-distillation and steam distillation from aromatic plants Sudeep Tandon Scientist Chemical Engineering Division CIMAP, Lucknow HISTORY Written records of herbal distillation are found as early as the first century A.D., and around 1000 A.D., the noted Arab physician and naturalist Ibn Sina also known as Avicenna described the distillation of rose oil from rose petals The ancient Arabian people began to study the chemical properties of essential oils & developed and refined the distillation process Europeans began producing essential oils in the 12th century 1 DISTILLATION ? A process in which a liquid or vapour mixture of two or more substances is separated into its component fractions of desired purity, by the application and removal of heat. In simple terms distillation of aromatic herbs implies vaporizing or liberating the oils from the trichomes / plant cell membranes of the herb in presence of high temperature and moisture and then cooling the vapour mixture to separate out the oil from water. It is the most popular widely used and cost effective method in use today for producing majority of the essential oils throughout the world Distillation is an art and not just a “Chemical" process that is reliant upon many factors for successful quality oil production. BASIC SCIENTIFIC PRINCIPLES INVOLVED IN THE PROCESS To convert any liquid into a vapour we have to apply energy in form of heat called as latent heat of vaporization A liquid always boils at the temperature at which its vapour pressure equals the atmospheric / surrounding pressure For two immiscible liquids the total vapour pressure of the mixture is always equal to the sum of their partial pressures The composition of the mixture will be determined by the concentration of the individual components into its partial pressure As known the boiling point of most essential oil components exceeds that of water and generally lies between 150 – 300oC 2 If a sample of an essential oil having a component ‘A’ having boiling point for example 190oC and the boiling point of the water is 100oC.