Functional Analysis of Somatic Mutations Affecting Receptor Tyrosine Kinase Family in Metastatic Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
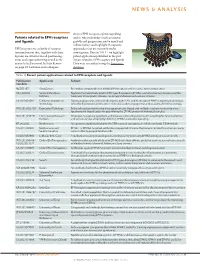
Patents Related to EPH Receptors and Ligands
NEWS & ANALYSIS discuss EPH receptor–ephrin signalling Patents related to EPH receptors and its role in disorders such as tumour and ligands growth and progression, nerve injury and inflammation, and highlight therapeutic EPH receptors are a family of receptor approaches that are currently under tyrosine kinases that, together with their investigation. Here in TABLE 1 we highlight ligands, are involved in cell positioning, patent applications published in the past tissue and organ patterning as well as the 3 years related to EPH receptors and ligands. control of cell survival. In their Review Data were researched using the Espacenet on page 39, Lackman and colleagues database. Table 1 | Recent patent applications related to EPH receptors and ligands Nature Reviews | Drug Discovery Publication Applicants Subject numbers NZ 581397 AstraZeneca Pyrimidine compounds that inhibit EPH receptors and are useful for treating cancer HK 1108702 Sanford-Burnham Peptides that selectively bind to EPH type-B receptors (EPHBs); useful for tumour imaging and the Institute treatment of neoplastic disease, neurological disease and vascular disease US 2013091591 California Institute of During angiogenesis, arterial cells express ephrin B2, and its receptor EPHB4 is expressed on venous Technology cells; this distinction can be used in methods to alter angiogenesis and to assess the effect of drugs WO 2013052710 Expression Pathology Selected reaction monitoring mass spectrometry-based and multiple reaction monitoring mass spectrometry-based assays for quantifying -

Human FLT4 / VEGFR3 ELISA Kit (ARG82047)
Product datasheet [email protected] ARG82047 Package: 96 wells Human FLT4 / VEGFR3 ELISA Kit Store at: 4°C Summary Product Description Human FLT4 / VEGFR3 ELISA Kit is an Enzyme Immunoassay kit for the quantification of Human FLT4 / VEGFR3 in serum, plasma and cell culture supernatants. Tested Reactivity Hu Tested Application ELISA Target Name FLT4 / VEGFR3 Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm. Sensitivity 78 pg/ml Sample Type Serum, plasma and cell culture supernatants. Standard Range 156 - 10000 pg/ml Sample Volume 100 µl Alternate Names FLT-4; FLT41; Vascular endothelial growth factor receptor 3; VEGFR3; VEGFR-3; PCL; Tyrosine-protein kinase receptor FLT4; LMPH1A; EC 2.7.10.1; Fms-like tyrosine kinase 4 Application Instructions Assay Time 4.5 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C. Keep microplate wells sealed in a dry bag with desiccants. Do not expose test reagents to heat, sun or strong light during storage and usage. Please refer to the product user manual for detail temperatures of the components. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol FLT4 Gene Full Name fms-related tyrosine kinase 4 Background This gene encodes a tyrosine kinase receptor for vascular endothelial growth factors C and D. The protein is thought to be involved in lymphangiogenesis and maintenance of the lymphatic endothelium. Mutations in this gene cause hereditary lymphedema type IA. [provided by RefSeq, Jul 2008] Function Tyrosine-protein kinase that acts as a cell-surface receptor for VEGFC and VEGFD, and plays an essential role in adult lymphangiogenesis and in the development of the vascular network and the cardiovascular system during embryonic development. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

A Review of VEGF/VEGFR-Targeted Therapeutics for Recurrent Glioblastoma
414 Original Article A Review of VEGF/VEGFR-Targeted Therapeutics for Recurrent Glioblastoma David A. Reardon, MDa,b; Scott Turner, MDc; Katherine B. Peters, MD, PhDc; Annick Desjardins, MDc; Sridharan Gururangan, MDa,b; John H. Sampson, MD, PhDa; Roger E. McLendon, MDd; James E. Herndon II, PhDe; Lee W. Jones, PhDf; John P. Kirkpatrick, MD, PhDf; Allan H. Friedman, MDa; James J. Vredenburgh, MDc; Darell D. Bigner, MD, PhDd; and Henry S. Friedman, MDa,b; Durham, North Carolina Key Words ability effect of VEGF inhibitors, the Radiologic Assessment in Neu- Glioblastoma, angiogenesis, vascular endothelial growth factor, ro-Oncology (RANO) criteria were recently implemented to bet- malignant glioma ter assess response among patients with glioblastoma. Although bevacizumab improves survival and quality of life, eventual tumor progression is the norm. Better understanding of resistance mech- Abstract anisms to VEGF inhibitors and identification of effective therapy Glioblastoma, the most common primary malignant brain tumor after bevacizumab progression are currently a critical need for pa- among adults, is a highly angiogenic and deadly tumor. Angiogen- tients with glioblastoma. (JNCCN 2011;9:414–427) esis in glioblastoma, driven by hypoxia-dependent and indepen- dent mechanisms, is primarily mediated by vascular endothelial growth factor (VEGF), and generates blood vessels with distinctive features. The outcome for patients with recurrent glioblastoma is poor because of ineffective therapies. However, recent encourag- ing rates of radiographic response and progression-free survival, Malignant gliomas, including the most common sub- and adequate safety, led the FDA to grant accelerated approval of type of glioblastoma, are rapidly growing destructive tu- bevacizumab, a humanized monoclonal antibody against VEGF, for mors that extensively invade locally but rarely metasta- the treatment of recurrent glioblastoma in May 2009. -

PTPRA Phosphatase Regulates GDNF-Dependent RET Signaling and Inhibits the RET Mutant MEN2A Oncogenic Potential
Journal Pre-proof PTPRA phosphatase regulates GDNF-dependent RET signaling and inhibits the RET mutant MEN2A oncogenic potential Leena Yadav, Elina Pietilä, Tiina Öhman, Xiaonan Liu, Arun K. Mahato, Yulia Sidorova, Kaisa Lehti, Mart Saarma, Markku Varjosalo PII: S2589-0042(20)30055-9 DOI: https://doi.org/10.1016/j.isci.2020.100871 Reference: ISCI 100871 To appear in: ISCIENCE Received Date: 3 August 2019 Revised Date: 15 January 2020 Accepted Date: 26 January 2020 Please cite this article as: Yadav, L., Pietilä, E., Öhman, T., Liu, X., Mahato, A.K., Sidorova, Y., Lehti, K., Saarma, M., Varjosalo, M., PTPRA phosphatase regulates GDNF-dependent RET signaling and inhibits the RET mutant MEN2A oncogenic potential, ISCIENCE (2020), doi: https://doi.org/10.1016/ j.isci.2020.100871. This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. © 2020 Growth factor RET PTPRA Cell surface Ras P P Complex formation RAF MEK ERK Growth Nucleus Proliferation Gene expression Migration 1 PTPRA phosphatase regulates GDNF-dependent RET signaling and inhibits the RET mutant MEN2A oncogenic potential Authors Leena Yadav 1, Elina Pietilä 3# , Tiina Öhman 1# , Xiaonan Liu 1, Arun K. -

Elephantid Genomes Reveal the Molecular Bases of Woolly Mammoth Adaptations to the Arctic
Article Elephantid Genomes Reveal the Molecular Bases of Woolly Mammoth Adaptations to the Arctic Graphical Abstract Authors Vincent J. Lynch, Oscar C. Bedoya-Reina, Aakrosh Ratan, ..., George H. Perry, Webb Miller, Stephan C. Schuster Correspondence [email protected] (V.J.L.), [email protected] (W.M.) In Brief Lynch et al. sequence complete genomes from three Asian elephants and two woolly mammoths and identify amino acid changes unique to woolly mammoths. Woolly-mammoth-specific amino acid changes underlie cold- adapted traits in mammoths, including small ears, thick fur, and altered temperature sensation. Highlights d Complete genomes of three Asian elephants and two woolly mammoths were sequenced d Mammoth-specific amino acid changes were found in 1,642 protein-coding genes d Genes with mammoth-specific changes are associated with adaptation to extreme cold d An amino acid change in TRPV3 may have altered temperature sensation in mammoths Lynch et al., 2015, Cell Reports 12, 217–228 July 14, 2015 ª2015 The Authors http://dx.doi.org/10.1016/j.celrep.2015.06.027 Cell Reports Article Elephantid Genomes Reveal the Molecular Bases of Woolly Mammoth Adaptations to the Arctic Vincent J. Lynch,1,* Oscar C. Bedoya-Reina,2,4 Aakrosh Ratan,2,5 Michael Sulak,1 Daniela I. Drautz-Moses,2,6 George H. Perry,3 Webb Miller,2,* and Stephan C. Schuster2,6 1Department of Human Genetics, The University of Chicago, 920 East 58th Street, CLSC 319C, Chicago, IL 60637, USA 2Center for Comparative Genomics and Bioinformatics, Pennsylvania State University, 506B -

Supplemental Tables4.Pdf
Yano_Supplemental_Table_S4 Gene ontology – Biological process 1 of 9 Fold List Pop Pop GO Term Count % PValue Bonferroni Benjamini FDR Genes Total Hits Total Enrichment DLC1, CADM1, NELL2, CLSTN1, PCDHGA8, CTNNB1, NRCAM, APP, CNTNAP2, FERT2, RAPGEF1, PTPRM, MPDZ, SDK1, PCDH9, PTPRS, VEZT, NRXN1, MYH9, GO:0007155~cell CTNNA2, NCAM1, NCAM2, DDR1, LSAMP, CNTN1, 50 5.61 2.14E-08 510 311 7436 2.34 4.50E-05 4.50E-05 3.70E-05 adhesion ROR2, VCAN, DST, LIMS1, TNC, ASTN1, CTNND2, CTNND1, CDH2, NEO1, CDH4, CD24A, FAT3, PVRL3, TRO, TTYH1, MLLT4, LPP, NLGN1, PCDH19, LAMA1, ITGA9, CDH13, CDON, PSPC1 DLC1, CADM1, NELL2, CLSTN1, PCDHGA8, CTNNB1, NRCAM, APP, CNTNAP2, FERT2, RAPGEF1, PTPRM, MPDZ, SDK1, PCDH9, PTPRS, VEZT, NRXN1, MYH9, GO:0022610~biological CTNNA2, NCAM1, NCAM2, DDR1, LSAMP, CNTN1, 50 5.61 2.14E-08 510 311 7436 2.34 4.50E-05 4.50E-05 3.70E-05 adhesion ROR2, VCAN, DST, LIMS1, TNC, ASTN1, CTNND2, CTNND1, CDH2, NEO1, CDH4, CD24A, FAT3, PVRL3, TRO, TTYH1, MLLT4, LPP, NLGN1, PCDH19, LAMA1, ITGA9, CDH13, CDON, PSPC1 DCC, ENAH, PLXNA2, CAPZA2, ATP5B, ASTN1, PAX6, ZEB2, CDH2, CDH4, GLI3, CD24A, EPHB1, NRCAM, GO:0006928~cell CTTNBP2, EDNRB, APP, PTK2, ETV1, CLASP2, STRBP, 36 4.04 3.46E-07 510 205 7436 2.56 7.28E-04 3.64E-04 5.98E-04 motion NRG1, DCLK1, PLAT, SGPL1, TGFBR1, EVL, MYH9, YWHAE, NCKAP1, CTNNA2, SEMA6A, EPHA4, NDEL1, FYN, LRP6 PLXNA2, ADCY5, PAX6, GLI3, CTNNB1, LPHN2, EDNRB, LPHN3, APP, CSNK2A1, GPR45, NRG1, RAPGEF1, WWOX, SGPL1, TLE4, SPEN, NCAM1, DDR1, GRB10, GRM3, GNAQ, HIPK1, GNB1, HIPK2, PYGO1, GO:0007166~cell RNF138, ROR2, CNTN1, -

Ptk7-Deficient Mice Have Decreased Hematopoietic Stem Cell Pools As a Result of Deregulated Proliferation and Migration
Ptk7-Deficient Mice Have Decreased Hematopoietic Stem Cell Pools as a Result of Deregulated Proliferation and Migration This information is current as Anne-Catherine Lhoumeau, Marie-Laure Arcangeli, Maria of September 24, 2021. De Grandis, Marilyn Giordano, Jean-Christophe Orsoni, Frédérique Lembo, Florence Bardin, Sylvie Marchetto, Michel Aurrand-Lions and Jean-Paul Borg J Immunol 2016; 196:4367-4377; Prepublished online 18 April 2016; Downloaded from doi: 10.4049/jimmunol.1500680 http://www.jimmunol.org/content/196/10/4367 Supplementary http://www.jimmunol.org/content/suppl/2016/04/16/jimmunol.150068 http://www.jimmunol.org/ Material 0.DCSupplemental References This article cites 55 articles, 24 of which you can access for free at: http://www.jimmunol.org/content/196/10/4367.full#ref-list-1 Why The JI? Submit online. by guest on September 24, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Rat FLT4 / VEGFR3 ELISA Kit (ARG82090)
Product datasheet [email protected] ARG82090 Package: 96 wells Rat FLT4 / VEGFR3 ELISA Kit Store at: 4°C Component Cat. No. Component Name Package Temp ARG82090-001 Antibody-coated 8 X 12 strips 4°C. Unused strips microplate should be sealed tightly in the air-tight pouch. ARG82090-002 Standard 2 X 10 ng/vial 4°C ARG82090-003 Standard/Sample 30 ml (Ready to use) 4°C diluent ARG82090-004 Antibody conjugate 1 vial (100 µl) 4°C concentrate (100X) ARG82090-005 Antibody diluent 12 ml (Ready to use) 4°C buffer ARG82090-006 HRP-Streptavidin 1 vial (100 µl) 4°C concentrate (100X) ARG82090-007 HRP-Streptavidin 12 ml (Ready to use) 4°C diluent buffer ARG82090-008 25X Wash buffer 20 ml 4°C ARG82090-009 TMB substrate 10 ml (Ready to use) 4°C (Protect from light) ARG82090-010 STOP solution 10 ml (Ready to use) 4°C ARG82090-011 Plate sealer 4 strips Room temperature Summary Product Description ARG82090 Rat FLT4 / VEGFR3 ELISA Kit is an Enzyme Immunoassay kit for the quantification of Rat FLT4 / VEGFR3 in serum and cell culture supernatants. Tested Reactivity Rat Tested Application ELISA Specificity There is no detectable cross-reactivity with other relevant proteins. Target Name FLT4 / VEGFR3 Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm. Sensitivity 78 pg/ml Sample Type Serum and cell culture supernatants. Standard Range 156 - 10000 pg/ml Sample Volume 100 µl www.arigobio.com 1/3 Precision Intra-Assay CV: 5.2%; Inter-Assay CV: 6.2% Alternate Names FLT-4; FLT41; Vascular endothelial growth factor receptor 3; VEGFR3; VEGFR-3; PCL; Tyrosine-protein kinase receptor FLT4; LMPH1A; EC 2.7.10.1; Fms-like tyrosine kinase 4 Application Instructions Assay Time ~ 5 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C. -

Mesenchymal–Epithelial Interactions Involving Epiregulin in Tuberous Sclerosis Complex Hamartomas
Mesenchymal–epithelial interactions involving epiregulin in tuberous sclerosis complex hamartomas Shaowei Li*, Fumiko Takeuchi*, Ji-an Wang*, Qingyuan Fan*, Toshi Komurasaki†, Eric M. Billings‡, Gustavo Pacheco-Rodriguez‡, Joel Moss‡, and Thomas N. Darling*§ *Department of Dermatology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814-4712; †Molecular Biology Laboratory, Molecular and Pharmacology Laboratories, Taisho Pharmaceutical Co., Ltd., 430-1 Yoshino-cho, Saitma-shi, Saitama 331-9530, Japan; and ‡Translational Medicine Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Building 10, Room 6D05, MSC 1590, Bethesda, MD 20892-1590 Communicated by Martha Vaughan, National Institutes of Health, Bethesda, MD, December 31, 2007 (received for review November 30, 2007) Patients with tuberous sclerosis complex (TSC) develop hamarto- Like other hamartomas, those in TSC skin contain abnormal mas containing biallelic inactivating mutations in either TSC1 or numbers of several types of cells. In the dermis, there are TSC2, resulting in mammalian target of rapamycin (mTOR) activa- increased numbers of large stellate fibroblasts, capillaries, and tion. Hamartomas overgrow epithelial and mesenchymal cells in dermal dendritic cells (6–9). The epidermis is acanthotic (i.e., TSC skin. The pathogenetic mechanisms for these changes had not thickened from increased numbers of keratinocytes in the spi- been investigated, and the existence or location of cells with nous layer). Acanthosis is pronounced in PFs and variable in AFs biallelic mutations (‘‘two-hit’’ cells) was unclear. We compared TSC (7, 8). The epidermis of treated AFs, several months after argon skin hamartomas (angiofibromas and periungual fibromas) with or CO2 laser surgery, no longer appears acanthotic (10, 11). -

2020 Program Book
PROGRAM BOOK Note that TAGC was cancelled and held online with a different schedule and program. This document serves as a record of the original program designed for the in-person meeting. April 22–26, 2020 Gaylord National Resort & Convention Center Metro Washington, DC TABLE OF CONTENTS About the GSA ........................................................................................................................................................ 3 Conference Organizers ...........................................................................................................................................4 General Information ...............................................................................................................................................7 Mobile App ....................................................................................................................................................7 Registration, Badges, and Pre-ordered T-shirts .............................................................................................7 Oral Presenters: Speaker Ready Room - Camellia 4.......................................................................................7 Poster Sessions and Exhibits - Prince George’s Exhibition Hall ......................................................................7 GSA Central - Booth 520 ................................................................................................................................8 Internet Access ..............................................................................................................................................8