West2017.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vocabulario De Morfoloxía, Anatomía E Citoloxía Veterinaria
Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) Servizo de Normalización Lingüística Universidade de Santiago de Compostela COLECCIÓN VOCABULARIOS TEMÁTICOS N.º 4 SERVIZO DE NORMALIZACIÓN LINGÜÍSTICA Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) 2008 UNIVERSIDADE DE SANTIAGO DE COMPOSTELA VOCABULARIO de morfoloxía, anatomía e citoloxía veterinaria : (galego-español- inglés) / coordinador Xusto A. Rodríguez Río, Servizo de Normalización Lingüística ; autores Matilde Lombardero Fernández ... [et al.]. – Santiago de Compostela : Universidade de Santiago de Compostela, Servizo de Publicacións e Intercambio Científico, 2008. – 369 p. ; 21 cm. – (Vocabularios temáticos ; 4). - D.L. C 2458-2008. – ISBN 978-84-9887-018-3 1.Medicina �������������������������������������������������������������������������veterinaria-Diccionarios�������������������������������������������������. 2.Galego (Lingua)-Glosarios, vocabularios, etc. políglotas. I.Lombardero Fernández, Matilde. II.Rodríguez Rio, Xusto A. coord. III. Universidade de Santiago de Compostela. Servizo de Normalización Lingüística, coord. IV.Universidade de Santiago de Compostela. Servizo de Publicacións e Intercambio Científico, ed. V.Serie. 591.4(038)=699=60=20 Coordinador Xusto A. Rodríguez Río (Área de Terminoloxía. Servizo de Normalización Lingüística. Universidade de Santiago de Compostela) Autoras/res Matilde Lombardero Fernández (doutora en Veterinaria e profesora do Departamento de Anatomía e Produción Animal. -
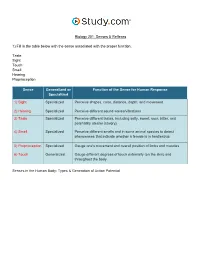
Senses & Reflexes in the Nervous System Visual Worksheet
Biology 201: Senses & Reflexes 1) Fill in the table below with the sense associated with the proper function. Taste Sight Touch Smell Hearing Proprioception Sense Generalized or Function of the Sense for Human Response Specialized 1) Sight Specialized Perceive shapes, color, distance, depth, and movement 2) Hearing Specialized Perceive different sound waves/vibrations 3) Taste Specialized Perceive different tastes, including salty, sweet, sour, bitter, and potentially umami (savory) 4) Smell Specialized Perceive different smells and in some animal species to detect pheromones that indicate whether a female is in heat/estrus 5) Proprioception Specialized Gauge one's movement and overall position of limbs and muscles 6) Touch Generalized Gauge different degrees of touch externally (on the skin) and throughout the body Senses in the Human Body: Types & Generation of Action Potential 2) Label the structures of the eye. Pupil Sclera Retina Lens Iris Cornea The Eye: Structure, Image Detection & Disorders 3) Label the chambers and structures of the eye below. Posterior chamber Muscle Retina Conjunctiva Cornea Sclera Optic nerve Anterior chamber Pupil Choroid layer Blood vessels Lens Iris Vitreous chamber The Eye: Structure, Image Detection & Disorders 4) Label the features of the visual pathway below. Optic chiasm Left cerebral hemisphere Pretectal nucleus Lateral geniculate nucleus of the thalamus Superior colliculus Visual cortex Right cerebral hemisphere The Eye: Structure, Image Detection & Disorders 5) Study the image of the section of the retina below. Label the neural layers. Bipolar cell Cone cell Neural layer Pigmented layer Rod cell Ganglion cell The Eye: Structure, Image Detection & Disorders 6) Label the structures of the nose below. -

Ear Infections in Children
U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES ∙ National Institutes of Health NIDCD Fact Sheet | Hearing and Balance Ear Infections in Children What is an ear infection? How can I tell if my child has an ear infection? An ear infection is an inflammation of the middle ear, usually caused by bacteria, that occurs when fluid builds Most ear infections happen to children before they’ve up behind the eardrum. Anyone can get an ear infection, learned how to talk. If your child isn’t old enough to say but children get them more often than adults. Five out of “My ear hurts,” here are a few things to look for: six children will have at least one ear infection by their third } Tugging or pulling at the ear(s) birthday. In fact, ear infections are the most common reason parents bring their child to a doctor. The scientific name for } Fussiness and crying an ear infection is otitis media (OM). } Trouble sleeping What are the symptoms of an } Fever (especially in infants and younger children) ear infection? } Fluid draining from the ear } Clumsiness or problems with balance There are three main types of ear infections. Each has a different combination of symptoms. } Trouble hearing or responding to quiet sounds. } Acute otitis media (AOM) is the most common ear What causes an ear infection? infection. Parts of the middle ear are infected and swollen and fluid is trapped behind the eardrum. This An ear infection usually is caused by bacteria and often causes pain in the ear—commonly called an earache. -

Patient Information – Ear Surgery Instructions
The Oregon Clinic, Plaza ENT Division 5050 NE Hoyt #655, Portland, OR 97213 Phone: 5034882400 Fax: 5032310121 Patient Information – Ear Surgery Instructions Pre- and Post-operative Instructions for Ear Surgery (not including ear tubes) Before Surgery: Many ear surgeries involve manipulation of the eardrum (tympanic membrane), and some require the removal of bone to facilitate the treatment of your ear disease. As with any operation, infection, scarring, and blood clot formation (hematoma) are possible. The facial nerve is at risk for injury or temporary weakness during any ear surgery. Dizziness following surgery may be expected. Hearing loss or ringing in the ear (tinnitus) may be more pronounced. Taste disturbance is not uncommon in certain ear surgeries for a few weeks following surgery and, in a few instances, could be prolonged or permanent. An incision may be made behind your ear, on your earlobe, or behind the pointed cartilage in front of your ear (the tragus). These areas normally heal without problems or obvious scars. Hair around the ear may or may not be shaved. Flying is usually permitted one month after surgery. Swimming may be allowed six weeks after surgery, but check with your doctor first before resuming swimming or other water sports. If your work is not strenuous and depending upon the type of surgery you’ve had, you may return to work 3 to 4 days from the date of surgery. Generally, you will be seen about 2-3 weeks after surgery. This gives your eardrum time to heal before we see you back. Pre-operative Instructions: 1. -

Basic Anatomy and Physiology of the Ear 11
1 BASIC ANATOMY AND PHYSIOLOGY OF THE EAR J. Irwin Introduction The ear is a small, complex series of interlinked structures that are involved in both maintenance of normal balance and the sense of hearing. In order to hear, the ear collects the sound waves that arrive as pressure changes in air and converts these into neurochemical impulses that travel along the cochlear- vestibular nerve to the brain. There are both active and passive mechanisms involved in this process.The prime function of the vestibular system is to detect and compensate for movement. This includes the ability to maintain optic fix- ation despite movement and to initiate muscle reflexes to maintain balance. For the purposes of describing structure and function the ear is usually split into four distinct parts. These are the outer ear, the middle ear and the audi- tory and vestibular parts of the inner ear (Figure 1.1). The outer ear This is sometimes known as the external ear and consists of the ear that is visible on the side of the head (the pinna), the external auditory meatus (ear hole) and the ear canal (external auditory canal) that leads to the eardrum (or tympanic membrane). The tympanic membrane has three layers and the outer layer is usually included as part of the outer ear. THE PINNA This is, for the most part, a piece of cartilage covered by skin (Figure 1.2). There is also a fatty earlobe in most people. The skin covering the cartilage is Infection and Hearing Impairment. Edited by V.E. Newton and P.J. -

Intraoperative Neuromonitoring Techniques in the Surgical Management of Acoustic Neuromas
Neurosurg Focus 33 (3):E6, 2012 Intraoperative neuromonitoring techniques in the surgical management of acoustic neuromas TAEMIN OH, B.A.,1 DANIEL T. NAGASAWA, M.D.,1 BRENDAN M. FONG, B.S.,1 ANDY TRANG, B.S.,1 QUINtoN GOPEN, M.D.,3 ANDREW T. PARSA, M.D., PH.D.,2 AND ISAAC YANG, M.D.1,4 Departments of 1Neurosurgery and 3Otolaryngology ENT, David Geffen School of Medicine, University of California, Los Angeles; 4UCLA Jonsson Comprehensive Cancer Center, University of California, Los Angeles; and 2Department of Neurosurgery, UCSF School of Medicine, University of California, San Francisco, California Unfavorable outcomes such as facial paralysis and deafness were once unfortunate probable complications following resection of acoustic neuromas. However, the implementation of intraoperative neuromonitoring during acoustic neuroma surgery has demonstrated placing more emphasis on quality of life and preserving neurological function. A modern review demonstrates a great degree of recent success in this regard. In facial nerve monitoring, the use of modern electromyography along with improvements in microneurosurgery has significantly improved preservation. Recent studies have evaluated the use of video monitoring as an adjunctive tool to further improve outcomes for patients undergoing surgery. Vestibulocochlear nerve monitoring has also been extensively studied, with the most popular techniques including brainstem auditory evoked potential monitoring, electrocochleography, and direct compound nerve action potential monitoring. Among them, direct recording remains the most promising and preferred monitoring method for functional acoustic preservation. However, when compared with postoperative facial nerve function, the hearing preservation is only maintained at a lower rate. Here, the authors analyze the major intraoperative neuromonitoring techniques available for acoustic neuroma resection. -

Reconstruction of Acquired Pinna Defects
Reconstruction of Acquired Pinna Defects Dissertation submitted to THE TAMILNADU Dr. M. G. R. MEDICAL UNIVERSITY In partial fulfillment of the regulations for the award of the degree of MCh (PLASTIC SURGERY) BRANCH III THE TAMIL NADU DR. MGR. MEDICAL UNIVERSITY CHENNAI. AUGUST 2008 DECLARATION I solemnly declare that this dissertation “RECONSTRUCTION OF ACQUIRED PINNA DEFECTS” was prepared by me in the Department of Plastic, Reconstructive and Maxillofacial Surgery, Madras Medical College and Government General Hospital, Chennai under the guidance and supervision of Professor & HOD Department of Plastic, Reconstructive and Maxillofacial Surgery, Madras Medical College and Government General Hospital, Chennai between 2005 and 2008. This dissertation is submitted to the TamilNadu Dr. MGR Medical University, Chennai in partial fulfilment of the University requirements for the award of degree of MCh Plastic Surgery. Place: Chennai Date : CERTIFICATE This is to certify that this dissertation entitled “ RECONSTRUCTION OF ACQUIRED PINNA DEFECTS” is a bonafide record of the research work done by Dr. D.VINOTH KUMAR for the award of MCh Plastic Surgery, under the supervision of Professor & HOD, Plastic Surgery Madras Medical College and Government General Hospital, Chennai between 2005 and 2008. I also certify that this dissertation is the result of the independent work done by candidate. Professor and Head of the Department Dean Plastic, Reconstructive and Maxillofacial Surgery Madras Medical College and Madras Medical College Government General Hospital Chennai – 600 003. Chennai – 600 003. ACKNOWLEDGEMENT I am thankful to the Dean, Madras Medical College and Government General Hospital, Chennai for permitting me to carry out this study. I am very grateful to my teacher and guide Professor V Alamelu M.S., Mch, FICS, FMMS Professor & HOD, Plastic Reconstructive and Maxillofacial Surgery, Madras Medical College, Chennai for her expert guidance and help without which this study would not have been possible. -

Índice De Denominacións Españolas
VOCABULARIO Índice de denominacións españolas 255 VOCABULARIO 256 VOCABULARIO agente tensioactivo pulmonar, 2441 A agranulocito, 32 abaxial, 3 agujero aórtico, 1317 abertura pupilar, 6 agujero de la vena cava, 1178 abierto de atrás, 4 agujero dental inferior, 1179 abierto de delante, 5 agujero magno, 1182 ablación, 1717 agujero mandibular, 1179 abomaso, 7 agujero mentoniano, 1180 acetábulo, 10 agujero obturado, 1181 ácido biliar, 11 agujero occipital, 1182 ácido desoxirribonucleico, 12 agujero oval, 1183 ácido desoxirribonucleico agujero sacro, 1184 nucleosómico, 28 agujero vertebral, 1185 ácido nucleico, 13 aire, 1560 ácido ribonucleico, 14 ala, 1 ácido ribonucleico mensajero, 167 ala de la nariz, 2 ácido ribonucleico ribosómico, 168 alantoamnios, 33 acino hepático, 15 alantoides, 34 acorne, 16 albardado, 35 acostarse, 850 albugínea, 2574 acromático, 17 aldosterona, 36 acromatina, 18 almohadilla, 38 acromion, 19 almohadilla carpiana, 39 acrosoma, 20 almohadilla córnea, 40 ACTH, 1335 almohadilla dental, 41 actina, 21 almohadilla dentaria, 41 actina F, 22 almohadilla digital, 42 actina G, 23 almohadilla metacarpiana, 43 actitud, 24 almohadilla metatarsiana, 44 acueducto cerebral, 25 almohadilla tarsiana, 45 acueducto de Silvio, 25 alocórtex, 46 acueducto mesencefálico, 25 alto de cola, 2260 adamantoblasto, 59 altura a la punta de la espalda, 56 adenohipófisis, 26 altura anterior de la espalda, 56 ADH, 1336 altura del esternón, 47 adipocito, 27 altura del pecho, 48 ADN, 12 altura del tórax, 48 ADN nucleosómico, 28 alunarado, 49 ADNn, 28 -

The Sensory System
U.S. ARMY MEDICAL DEPARTMENT CENTER AND SCHOOL FORT SAM HOUSTON, TEXAS 78234-6100 THE SENSORY SYSTEM SUBCOURSE MD0582 EDITION 100 DEVELOPMENT This subcourse is approved for resident and correspondence course instruction. It reflects the current thought of the Academy of Health Sciences and conforms to printed Department of the Army doctrine as closely as currently possible. Development and progress render such doctrine continuously subject to change. ADMINISTRATION Students who desire credit hours for this correspondence subcourse must enroll in the subcourse. Application for enrollment should be made at the Internet website: http://www.atrrs.army.mil. You can access the course catalog in the upper right corner. Enter School Code 555 for medical correspondence courses. Copy down the course number and title. To apply for enrollment, return to the main ATRRS screen and scroll down the right side for ATRRS Channels. Click on SELF DEVELOPMENT to open the application; then follow the on-screen instructions. For comments or questions regarding enrollment, student records, or examination shipments, contact the Nonresident Instruction Branch at DSN 471-5877, commercial (210) 221-5877, toll-free 1-800-344-2380; fax: 210-221-4012 or DSN 471-4012, e-mail [email protected], or write to: NONRESIDENT INSTRUCTION BRANCH AMEDDC&S ATTN: MCCS-HSN 2105 11TH STREET SUITE 4191 FORT SAM HOUSTON TX 78234-5064 Be sure your social security number is on all correspondence sent to the Academy of Health Sciences. CLARIFICATION OF TERMINOLOGY When used in this publication, words such as "he," "him," "his," and "men" 'are intended to include both the masculine and feminine genders, unless specifically stated otherwise or when obvious in context. -

Systematic Review and Comparative
Revista de la Facultad de Medicina Journal of the Faculty of Medicine Rev. Fac. Med. 2020Año 72, Vol. 68, No. 1 Faculty of Medicine Editorial Committee Franklin Escobar Córdoba. MD.MPF.PhD. Universidad Nacional de Colombia. Colombia. Javier Eslava Schmalbach. MD.MSc.PhD. Universidad Nacional de Colombia. Colombia. Lisieux Elaine de Borba Telles MD. MPF. PhD. Universidade Federal do Rio Grande do Sul. Brazil. Adelaida Restrepo PhD. Arizona State University. USA. Eduardo De La Peña de Torres PhD. Consejo Superior de Investigaciones Científicas. España. Fernando Sánchez-Santed MD. Universidad de Almería. España. Gustavo C. Román MD. University of Texas at San Antonio. USA. Jorge E. Tolosa MD.MSCE. Oregon Health & Science University. USA. Jorge Óscar Folino MD. MPF. PhD. Universidad Nacional de La Plata. Argentina. Julio A. Chalela MD. Medical University of South Carolina. USA. Sergio Javier Villaseñor Bayardo MD. PhD. Universidad de Guadalajara. México. Cecilia Algarin MD., Universidad de Chile. Lilia María Sánchez MD., Université de Montréal. Claudia Rosario Portilla Ramírez PhD.(c)., Universidad de Barcelona. Marco Tulio de Mello MD. PhD., Universidade Federal de Sao Paulo. Dalva Poyares MD. PhD., Universidade Federal de São Paulo. Marcos German Mora González, PhD., Universidad de Chile Eduardo José Pedrero-Pérez, MSc. PhD., Instituto de Adicciones, Madrid Salud. María Angélica Martinez-Tagle MSc. PhD., Universidad de Chile. Emilia Chirveches-Pérez, PhD., Consorci Hospitalari de Vic María Dolores Gil Llario, PhD., Universitat de València Fernando Jaén Águila, MD, MSc., Hospital Virgen de las Nieves, Granada. María Isabel Izquierdo Macián, MD., Universidad de Valencia. Guillermo Felipe López Sánchez, MSc, PhD., Universidad de Murcia. Martine Bonnaure-Mallet PhD., Université de Rennes. -

Fat Graft Myringoplasty
64 Pomeroy St., Suite B, Cortland, NY 13045 607-662-4521 tel 607-662-4523 fax Surgeon – Dr. Manoj Kumar Date of Procedure: Fat Graft Myringoplasty Definition Myringo = tympanic membrane Plasty = a surgical procedure for the repair, restoration, or replacement (as by a prosthesis) of a part of the body This procedure involves repairing a small hole in the eardrum with a small piece of fat taken from the earlobe. Purpose of Procedure The purpose of this procedure is to restore a normally functioning eardrum. Holes in the eardrum may be due to infections that have drained through the eardrum, a traumatic injury, or a persistent opening following ear tube removal. Preparation As with any procedure in which anesthesia is administered, you will be asked not to eat or drink anything after midnight on the evening prior to your surgery. You may brush your teeth in the morning but not swallow the water. If you are on medications that must be taken, you will have discussed this with us and/or the anesthesiologist and instructions will have been given to you. The procedure will not be performed if you are currently taking, or have recently taken any medication that may interfere with your ability to clot your blood (“blood thinners, aspirin, anti-inflammatory medicines, etc...”). We will have reviewed all of your current medications with you during the pre-operative / pre-procedure consultation. You are obligated to inform us if anything has changed (medication or otherwise) since your previous visit. Procedure The operation involves taking a small piece of fat from the earlobe through a small incision. -

Nervous System - Sensory Input
Nervous system - sensory input. Overview: Sensory neurons are designed to register certain chemical, physical, electrical or other stimulus. If this stimulus is strong enough, this sets off an action potential. This impulse is then transmitted to the appropriate part of the nervous system. Incidentally, the perception of “light”, “sound”, etc. is not determined by the “receptor”, but rather on how/where the signal winds up (mostly in the brain). As a silly example: suppose we could unplug nerves coming into the brain from the eyes. Instead, we plug in nerves coming from the ears. Now sound would be registered as “light”, but who knows what this would be interpreted as. There are 5 main categories of sensory receptors (actually, lots more, but these will do for now): Mechanoreceptors - mechanical changes (pressure, touch, etc.). Pain receptors - sense pain. Thermoreceptors - temperature receptors. Chemoreceptors - taste, smell, other chemical receptors. Electromagnetic receptors - light, other electromagnetic waves. [Notice that this list really has nothing to do with the proverbial “5 senses”]. Mechano, pain & thermo receptors. In the skin - a lot of these come together in the skin and form our sense of “touch” [Fig. 29.3A, p. 590]. Touch is actually made up of several different receptors. Pain - this is very important and causes a strong negative reaction. This helps us avoid damage. Leprosy is a disease which (among other things) can shut down pain receptors. Mechanoreceptors in the skin: Deep touch (pressure) - for example, what you feel when sitting down. Light touch (often assisted by hairs) - used to determine texture, air currents, etc. Thermorecptors: Separate receptors for hot and cold (separate) exists.