The Complete Chloroplast Genome Sequence of Gentiana Lawrencei Var
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Buy Allamanda Creeper, Pentalinon Luteum (Yellow) - Plants Online at Nurserylive | Best Plants at Lowest Price
Buy allamanda creeper, pentalinon luteum (yellow) - plants online at nurserylive | Best plants at lowest price Allamanda Creeper, Pentalinon luteum (Yellow) - Plants Allamanda is a genus of flowering plants in the dogbane family, Apocynaceae. They contain a white latex. Rating: Not Rated Yet Price Variant price modifier: Base price with tax Price with discount ?499 Salesprice with discount Sales price ?499 Sales price without tax ?499 Discount Tax amount Ask a question about this product Description With this purchase you will get: 01 Allamanda Creeper, Pentalinon luteum (Yellow) Plant Description for Allamanda Creeper, Pentalinon luteum (Yellow) Plant height: 15 - 23 inches (38 - 59 cm) Plant spread: 3 - 5 inches (7 - 13 cm) 1 / 3 Buy allamanda creeper, pentalinon luteum (yellow) - plants online at nurserylive | Best plants at lowest price As shrub Allamanda grows very thick and gets covered with yellow bell-shaped 3 to 4 inches long blooms in growing season. Although it does not have tendrils to climb but provided support can grow as vine too. Common name(s): Golden trumpet vine Flower colours: Yellow Bloom time: Spring, Summer, Fall, Winter Max reachable height: 4 to 6 ft Difficulty to grow: Easy Planting and care Allamanda is a bushy nature plant, the large-sized container is preferable as the plant gets heavy with the time. You may need to re-pot it every year. Regardless of size, make sure container has drainage holes at the bottom to ensure smooth drainage of water. Put stone or brick pieces on the holes. Sunlight: Full sun Soil: Soil pH 7.6 to 7.8 (mildly alkaline), Any good-quality potting mix. -

Coumarins and Iridoids from Crucianella Graeca, Cruciata Glabra, Cruciata Laevipes and Cruciata Pedemontana (Rubiaceae) Maya Iv
Coumarins and Iridoids from Crucianella graeca, Cruciata glabra, Cruciata laevipes and Cruciata pedemontana (Rubiaceae) Maya Iv. Mitova3, Mincho E. Anchevb, Stefan G. Panev3, Nedjalka V. Handjieva3 and Simeon S. Popov3 a Institute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Sofia 1113, Bulgaria h Institute of Botany, Bulgarian Academy of Sciences, Sofia 1113, Bulgaria Z. Naturforsch. 51c, 631-634 (1996); received May 23/July8 , 1996 Rubiaceae, Crucianella, Cruciata, Coumarins, Iridoids The coumarin and iridoid composition of Crucianella graeca, Cruciata glabra, Cruciata laevipes and Cruciata pedemontana has been studied. Daphnin and daphnetin glucoside do minated in C. glabra along with low concentrations of daphnetin, deacetylasperulosidic acid and scandoside. In C. laevipes and C. pedemontana were found the same coumarin glucosides along with six iridoid glucosides. In Crucianella graeca were found ten iridoid glucosides. Introduction Thirteen pure compounds (Fig. 1) were isolated and identified by 'H and 13C NMR spectra (Ta Crucianella L. and Cruciata Mill. (Rubiaceae) ble I) and comparisons with authentic samples as are represented each by three species in the Bul the coumarins, daphnin (1), daphnetin glucoside garian flora (Ancev, 1976, 1979). They are mor (2) and daphnetin (3) (Jevers et al., 1978) and the phologically well differentiated and all except iridoids, deacetylasperulosidic acid (4), scandoside Cruciata glabra (L.) Ehrend., do not suggest seri (5), asperuloside (6), asperulosidic acid (7), methyl ous taxonomic problems. The coumarins, scopo- ester of deacetylasperulosidic acid (8), daphyllo letin, umbelliferone and cruciatin and the iridoid side (9), geniposidic acid (10), 10-hydroxyloganin glucosides monotropein, asperuloside and au- (11), deacetylasperuloside (12) and iridoid V3 (13) cubin, were found in some Cruciata species (Bo (Boros and Stermitz, 1990; El-Naggar & Beal, risov, 1967, 1974; Borisov and Borisyuk, 1965; Bo 1980). -

FLORA from FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE of MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2
ISSN: 2601 – 6141, ISSN-L: 2601 – 6141 Acta Biologica Marisiensis 2018, 1(1): 60-70 ORIGINAL PAPER FLORA FROM FĂRĂGĂU AREA (MUREŞ COUNTY) AS POTENTIAL SOURCE OF MEDICINAL PLANTS Silvia OROIAN1*, Mihaela SĂMĂRGHIŢAN2 1Department of Pharmaceutical Botany, University of Medicine and Pharmacy of Tîrgu Mureş, Romania 2Mureş County Museum, Department of Natural Sciences, Tîrgu Mureş, Romania *Correspondence: Silvia OROIAN [email protected] Received: 2 July 2018; Accepted: 9 July 2018; Published: 15 July 2018 Abstract The aim of this study was to identify a potential source of medicinal plant from Transylvanian Plain. Also, the paper provides information about the hayfields floral richness, a great scientific value for Romania and Europe. The study of the flora was carried out in several stages: 2005-2008, 2013, 2017-2018. In the studied area, 397 taxa were identified, distributed in 82 families with therapeutic potential, represented by 164 medical taxa, 37 of them being in the European Pharmacopoeia 8.5. The study reveals that most plants contain: volatile oils (13.41%), tannins (12.19%), flavonoids (9.75%), mucilages (8.53%) etc. This plants can be used in the treatment of various human disorders: disorders of the digestive system, respiratory system, skin disorders, muscular and skeletal systems, genitourinary system, in gynaecological disorders, cardiovascular, and central nervous sistem disorders. In the study plants protected by law at European and national level were identified: Echium maculatum, Cephalaria radiata, Crambe tataria, Narcissus poeticus ssp. radiiflorus, Salvia nutans, Iris aphylla, Orchis morio, Orchis tridentata, Adonis vernalis, Dictamnus albus, Hammarbya paludosa etc. Keywords: Fărăgău, medicinal plants, human disease, Mureş County 1. -

Feasibility Study of Kailash Sacred Landscape
Kailash Sacred Landscape Conservation Initiative Feasability Assessment Report - Nepal Central Department of Botany Tribhuvan University, Kirtipur, Nepal June 2010 Contributors, Advisors, Consultants Core group contributors • Chaudhary, Ram P., Professor, Central Department of Botany, Tribhuvan University; National Coordinator, KSLCI-Nepal • Shrestha, Krishna K., Head, Central Department of Botany • Jha, Pramod K., Professor, Central Department of Botany • Bhatta, Kuber P., Consultant, Kailash Sacred Landscape Project, Nepal Contributors • Acharya, M., Department of Forest, Ministry of Forests and Soil Conservation (MFSC) • Bajracharya, B., International Centre for Integrated Mountain Development (ICIMOD) • Basnet, G., Independent Consultant, Environmental Anthropologist • Basnet, T., Tribhuvan University • Belbase, N., Legal expert • Bhatta, S., Department of National Park and Wildlife Conservation • Bhusal, Y. R. Secretary, Ministry of Forest and Soil Conservation • Das, A. N., Ministry of Forest and Soil Conservation • Ghimire, S. K., Tribhuvan University • Joshi, S. P., Ministry of Forest and Soil Conservation • Khanal, S., Independent Contributor • Maharjan, R., Department of Forest • Paudel, K. C., Department of Plant Resources • Rajbhandari, K.R., Expert, Plant Biodiversity • Rimal, S., Ministry of Forest and Soil Conservation • Sah, R.N., Department of Forest • Sharma, K., Department of Hydrology • Shrestha, S. M., Department of Forest • Siwakoti, M., Tribhuvan University • Upadhyaya, M.P., National Agricultural Research Council -

Download (592Kb)
This is an Open Access document downloaded from ORCA, Cardiff University's institutional repository: http://orca.cf.ac.uk/134581/ This is the author’s version of a work that was submitted to / accepted for publication. Citation for final published version: Raye, Lee 2020. The wild plants of Scotia Illustrata (1684). British & Irish Botany 2 (3) , pp. 240- 258. 10.33928/bib.2020.02.240 file Publishers page: http://dx.doi.org/10.33928/bib.2020.02.240 <http://dx.doi.org/10.33928/bib.2020.02.240> Please note: Changes made as a result of publishing processes such as copy-editing, formatting and page numbers may not be reflected in this version. For the definitive version of this publication, please refer to the published source. You are advised to consult the publisher’s version if you wish to cite this paper. This version is being made available in accordance with publisher policies. See http://orca.cf.ac.uk/policies.html for usage policies. Copyright and moral rights for publications made available in ORCA are retained by the copyright holders. British & Irish Botany 2(3): 240-258, 2020 The wild plants of Scotia Illustrata (1684) Lee Raye Cardiff University, Wales, UK Corresponding author: Lee Raye: [email protected] This pdf constitutes the Version of Record published on 31st August 2020 Abstract Scotia Illustrata was published in 1684 and contains a section (II:1) describing 662 ‘naturally occurring plants of Scotland’. This paper sets out to identify and discuss the species in the text. It was possible to identify 652 species from the text and 396 could be securely identified. -
Dwarf Thistle, Cirsi
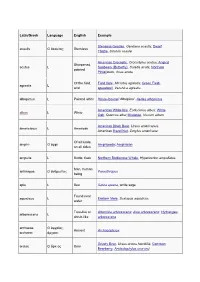
Latin/Greek Language English Example Stemless Gentian, Gentiana acaulis; Dwarf acaulis G ἄκαυλος Stemless Thistle, Cirsium acaule American Crocodile, Crocodylus acutus; Angled Sharpened, acutus L Sunbeam (Butterfly), Curetis acuta; Northern pointed Pintailduck, Anas acuta Of the field, Field Vole, Microtus agrestis; Green Field- agrestis L wild speedwell, Veronica agrestis albopictus L Painted white Hosta fortunei 'Albopicta', Aedes albopictus American White Ibis, Eudocimus albus; White albus L White Oak, Quercus alba; Mistletoe, Viscum album American Black Bear, Ursus americanus; americanus L American American Hazel Nut, Corylus americana Of all kinds, amphi- G ἀμφί Amphipoda; Amphibian on all sides ampulla L Bottle, flask Northern Bottlenose Whale, Hyperoodon ampullatus Man, human anthropos G ἄνθρωπος Paranthropus being apis L Bee Salvia apiana, white sage Found near aquaticus L Eastern Mole, Scalopus aquaticus water Tree-like or Artemisia arborescens; Aloe arborescens; Hydrangea arborescens L shrub-like arborescens archaeos, G ἀρχαῖος, Ancient Archaeopteryx archaeo- ἀρχαιο- Grizzly Bear, Ursus arctos horribilis; Common arctos G ἄρκτος Bear Bearberry, Arctostaphylos uva-ursi argentatus L Silvery Herring Gull, Larus argentatus arthron G ἄρθρον Joint Arthropoda arvensis L In the field Skylark, Alauda arvensis astron, astro-, G ἄστρον, Star Starfish (class), Asteroidea astero- ἀστρο-, ἀστερο- Acer palmatum 'Atropurpureum'; Berberis atropurpureum L Deep purple thunbergii f. atropurpurea Daphne odora 'Aureomarginata'; Taxus aureomarginata -

Caribbean Naturalist No
Caribbean Naturalist No. 13 2014 Natural Vegetation Groups and Canopy Chemical Markers in a Dry Subtropical Forest on Calcareous Substrate: The Vegetation of Mona Island, Puerto Rico Ernesto Medina, Eileen H. Helmer, Elvia Meléndez-Ackerman, and Humfredo Marcano-Vega The Caribbean Naturalist . ♦ A peer-reviewed and edited interdisciplinary natural history science journal with a re- gional focus on the Caribbean ( ISSN 2326-7119 [online]). ♦ Featuring research articles, notes, and research summaries on terrestrial, fresh-water, and marine organisms, and their habitats. The journal's versatility also extends to pub- lishing symposium proceedings or other collections of related papers as special issues. ♦ Focusing on field ecology, biology, behavior, biogeography, taxonomy, evolution, anatomy, physiology, geology, and related fields. Manuscripts on genetics, molecular biology, anthropology, etc., are welcome, especially if they provide natural history in- sights that are of interest to field scientists. ♦ Offers authors the option of publishing large maps, data tables, audio and video clips, and even powerpoint presentations as online supplemental files. ♦ Proposals for Special Issues are welcome. ♦ Arrangements for indexing through a wide range of services, including Web of Knowledge (includes Web of Science, Current Contents Connect, Biological Ab- stracts, BIOSIS Citation Index, BIOSIS Previews, CAB Abstracts), PROQUEST, SCOPUS, BIOBASE, EMBiology, Current Awareness in Biological Sciences (CABS), EBSCOHost, VINITI (All-Russian Institute of Scientific and Technical Information), FFAB (Fish, Fisheries, and Aquatic Biodiversity Worldwide), WOW (Waters and Oceans Worldwide), and Zoological Record, are being pursued. ♦ The journal staff is pleased to discuss ideas for manuscripts and to assist during all stages of manuscript preparation. The journal has a mandatory page charge to help defray a portion of the costs of publishing the manuscript. -

North American Rock Garden Society |
Bulletin of the American Rock Garden Society Volume 50 Number 4 Fall 1992 Cover: Gentiana paradoxa by Rob Proctor of Denver, Colorado Bulletin of the American Rock Garden Society Volume 50 Number 4 Fall 1992 Features Sorting out the Gentians, by Geoffrey Charlesworth 243 Fritillaries of Central Asia, by Josef Slegl 253 Trillium Rescue, by Don L. Jacobs 261 The Story of Fritillaria 'Martha Roderick', by W.H. de Goede 264 New Home for Rock Plants, by Elisabeth Sheldon 265 Eriogonums: Secret of the Dry Garden, by Irma Gourley 271 Preserving Rock Garden Specimens, by Karen Matthews 275 Spontaneity on the Rocks, by Panayoti Kelaidis 285 The Arctic Harebell, by J.S. DeSanto 291 Hunting for Red Helleborus niger, by Will McLewin 295 Departments Plant Portrait: Gentiana paradoxa 276 Awards 299 Books 305 Gentiana algida 242 Bulletin of the American Rock Garden Society Vol. 50(4) Sorting out the Gentians by Geoffrey Charlesworth 1 here are some genera in which tors. It is one of the hallmarks of a many of the species are considered good grower if a large patch can be good alpine plants. Androsace is such produced and maintained year after a genus, and we tend to dismiss the year, but the despair of most of us, who species that are not up to the highest have only occasionally seen a few small standard as not worth growing—for plants in our own gardens and then not instance, A. loctiflora or A. albana. It always with the astonishing color we is a mistake to make such odious associate with the species. -

Phylogeny and Phylogenetic Taxonomy of Dipsacales, with Special Reference to Sinadoxa and Tetradoxa (Adoxaceae)
PHYLOGENY AND PHYLOGENETIC TAXONOMY OF DIPSACALES, WITH SPECIAL REFERENCE TO SINADOXA AND TETRADOXA (ADOXACEAE) MICHAEL J. DONOGHUE,1 TORSTEN ERIKSSON,2 PATRICK A. REEVES,3 AND RICHARD G. OLMSTEAD 3 Abstract. To further clarify phylogenetic relationships within Dipsacales,we analyzed new and previously pub- lished rbcL sequences, alone and in combination with morphological data. We also examined relationships within Adoxaceae using rbcL and nuclear ribosomal internal transcribed spacer (ITS) sequences. We conclude from these analyses that Dipsacales comprise two major lineages:Adoxaceae and Caprifoliaceae (sensu Judd et al.,1994), which both contain elements of traditional Caprifoliaceae.Within Adoxaceae, the following relation- ships are strongly supported: (Viburnum (Sambucus (Sinadoxa (Tetradoxa, Adoxa)))). Combined analyses of C ap ri foliaceae yield the fo l l ow i n g : ( C ap ri folieae (Diervilleae (Linnaeeae (Morinaceae (Dipsacaceae (Triplostegia,Valerianaceae)))))). On the basis of these results we provide phylogenetic definitions for the names of several major clades. Within Adoxaceae, Adoxina refers to the clade including Sinadoxa, Tetradoxa, and Adoxa.This lineage is marked by herbaceous habit, reduction in the number of perianth parts,nectaries of mul- ticellular hairs on the perianth,and bifid stamens. The clade including Morinaceae,Valerianaceae, Triplostegia, and Dipsacaceae is here named Valerina. Probable synapomorphies include herbaceousness,presence of an epi- calyx (lost or modified in Valerianaceae), reduced endosperm,and distinctive chemistry, including production of monoterpenoids. The clade containing Valerina plus Linnaeeae we name Linnina. This lineage is distinguished by reduction to four (or fewer) stamens, by abortion of two of the three carpels,and possibly by supernumerary inflorescences bracts. Keywords: Adoxaceae, Caprifoliaceae, Dipsacales, ITS, morphological characters, phylogeny, phylogenetic taxonomy, phylogenetic nomenclature, rbcL, Sinadoxa, Tetradoxa. -

The Vascular Flora of Rarău Massif (Eastern Carpathians, Romania). Note Ii
Memoirs of the Scientific Sections of the Romanian Academy Tome XXXVI, 2013 BIOLOGY THE VASCULAR FLORA OF RARĂU MASSIF (EASTERN CARPATHIANS, ROMANIA). NOTE II ADRIAN OPREA1 and CULIŢĂ SÎRBU2 1 “Anastasie Fătu” Botanical Garden, Str. Dumbrava Roşie, nr. 7-9, 700522–Iaşi, Romania 2 University of Agricultural Sciences and Veterinary Medicine Iaşi, Faculty of Agriculture, Str. Mihail Sadoveanu, nr. 3, 700490–Iaşi, Romania Corresponding author: [email protected] This second part of the paper about the vascular flora of Rarău Massif listed approximately half of the whole number of the species registered by the authors in their field trips or already included in literature on the same area. Other taxa have been added to the initial list of plants, so that, the total number of taxa registered by the authors in Rarău Massif amount to 1443 taxa (1133 species and 310 subspecies, varieties and forms). There was signaled out the alien taxa on the surveyed area (18 species) and those dubious presence of some taxa for the same area (17 species). Also, there were listed all the vascular plants, protected by various laws or regulations, both internal or international, existing in Rarău (i.e. 189 taxa). Finally, there has been assessed the degree of wild flora conservation, using several indicators introduced in literature by Nowak, as they are: conservation indicator (C), threat conservation indicator) (CK), sozophytisation indicator (W), and conservation effectiveness indicator (E). Key words: Vascular flora, Rarău Massif, Romania, conservation indicators. 1. INTRODUCTION A comprehensive analysis of Rarău flora, in terms of plant diversity, taxonomic structure, biological, ecological and phytogeographic characteristics, as well as in terms of the richness in endemics, relict or threatened plant species was published in our previous note (see Oprea & Sîrbu 2012). -

704-717, 2011 Issn 1991-8178
Australian Journal of Basic and Applied Sciences, 5(6): 704-717, 2011 ISSN 1991-8178 Regeneration, Cardenolide and Flavonoid Production from In Vitro Cultures of Cynanchum acutum L. (Asclepiadaceae) 1Ahmed A. El-Bakry, 2Ezzat Genady, 1Safeyia M. Ghazi and 1Shams A. Rafat 1Botany Department, Faculty of Science, Helwan University, Cairo, Egypt. 2Pharmacognosy Department, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt. Summary: Callus was produced from hypocotyl sections of in vitro germinated seedlings of Cynanchum acutum on Murashige and Skoog media (1962) containing 0.5 - 4.0 mg/l a-Naphthalene acetic acid in combination with 0.2-1.0 mg/l Benzyl adenine. After 12 weeks in culture both fresh and dry weights (g) were significantly higher on 0.5 mg/l NAA and 0.2 mg/l BA. Cardiac glycoside concentration was highest (1.2 mg/g DW) on the same growth regulators combination. Flavonoids were highest (0.198 mg/g) on 0.5mg/l NAA and 1.0 mg/l BA. Twenty eight weeks old callus gave highest cardiac glycosides (1.3 mg/g) on media lacking BA and 0.5 mg/l NAA. Flavonoids concentration (0.27 mg/g) was highest on the same auxin concentration in the presence of 0.2 mg/l BA. Regeneration from callus cultures was obtained when 8-weeks old callus was subcultured on MS hormone free media for 6 weeks. Shoots were rooted on MS supplemented with 0.1 mg/l NAA and acclimatized in growth chamber. Adventitious shoots showed significantly higher CG (6.5 and 4.3 mg/g) than the wild plants (2.8), while regenerants gave comparable concentration (2.3) to the wild. -

Report of Rapid Biodiversity Assessments at Cenwanglaoshan Nature Reserve, Northwest Guangxi, China, 1999 and 2002
Report of Rapid Biodiversity Assessments at Cenwanglaoshan Nature Reserve, Northwest Guangxi, China, 1999 and 2002 Kadoorie Farm and Botanic Garden in collaboration with Guangxi Zhuang Autonomous Region Forestry Department Guangxi Forestry Survey and Planning Institute South China Institute of Botany South China Normal University Institute of Zoology, CAS March 2003 South China Forest Biodiversity Survey Report Series: No. 27 (Online Simplified Version) Report of Rapid Biodiversity Assessments at Cenwanglaoshan Nature Reserve, Northwest Guangxi, China, 1999 and 2002 Editors John R. Fellowes, Bosco P.L. Chan, Michael W.N. Lau, Ng Sai-Chit and Gloria L.P. Siu Contributors Kadoorie Farm and Botanic Garden: Gloria L.P. Siu (GS) Bosco P.L. Chan (BC) John R. Fellowes (JRF) Michael W.N. Lau (ML) Lee Kwok Shing (LKS) Ng Sai-Chit (NSC) Graham T. Reels (GTR) Roger C. Kendrick (RCK) Guangxi Zhuang Autonomous Region Forestry Department: Xu Zhihong (XZH) Pun Fulin (PFL) Xiao Ma (XM) Zhu Jindao (ZJD) Guangxi Forestry Survey and Planning Institute (Comprehensive Tan Wei Fu (TWF) Planning Branch): Huang Ziping (HZP) Guangxi Natural History Museum: Mo Yunming (MYM) Zhou Tianfu (ZTF) South China Institute of Botany: Chen Binghui (CBH) Huang Xiangxu (HXX) Wang Ruijiang (WRJ) South China Normal University: Li Zhenchang (LZC) Chen Xianglin (CXL) Institute of Zoology CAS (Beijing): Zhang Guoqing (ZGQ) Chen Deniu (CDN) Nanjing University: Chen Jianshou (CJS) Wang Songjie (WSJ) Xinyang Teachers’ College: Li Hongjing (LHJ) Voluntary specialist: Keith D.P. Wilson (KW) Background The present report details the findings of visits to Northwest Guangxi by members of Kadoorie Farm and Botanic Garden (KFBG) in Hong Kong and their colleagues, as part of KFBG's South China Biodiversity Conservation Programme.