Deep Learning for (Unsupervised) Clustering of DNA Sequences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Beta Diversity Patterns of Fish and Conservation Implications in The
A peer-reviewed open-access journal ZooKeys 817: 73–93 (2019)Beta diversity patterns of fish and conservation implications in... 73 doi: 10.3897/zookeys.817.29337 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research Beta diversity patterns of fish and conservation implications in the Luoxiao Mountains, China Jiajun Qin1,*, Xiongjun Liu2,3,*, Yang Xu1, Xiaoping Wu1,2,3, Shan Ouyang1 1 School of Life Sciences, Nanchang University, Nanchang 330031, China 2 Key Laboratory of Poyang Lake Environment and Resource Utilization, Ministry of Education, School of Environmental and Chemical Engi- neering, Nanchang University, Nanchang 330031, China 3 School of Resource, Environment and Chemical Engineering, Nanchang University, Nanchang 330031, China Corresponding author: Shan Ouyang ([email protected]); Xiaoping Wu ([email protected]) Academic editor: M.E. Bichuette | Received 27 August 2018 | Accepted 20 December 2018 | Published 15 January 2019 http://zoobank.org/9691CDA3-F24B-4CE6-BBE9-88195385A2E3 Citation: Qin J, Liu X, Xu Y, Wu X, Ouyang S (2019) Beta diversity patterns of fish and conservation implications in the Luoxiao Mountains, China. ZooKeys 817: 73–93. https://doi.org/10.3897/zookeys.817.29337 Abstract The Luoxiao Mountains play an important role in maintaining and supplementing the fish diversity of the Yangtze River Basin, which is also a biodiversity hotspot in China. However, fish biodiversity has declined rapidly in this area as the result of human activities and the consequent environmental changes. Beta diversity was a key concept for understanding the ecosystem function and biodiversity conservation. Beta diversity patterns are evaluated and important information provided for protection and management of fish biodiversity in the Luoxiao Mountains. -

Family-Cyprinidae-Gobioninae-PDF
SUBFAMILY Gobioninae Bleeker, 1863 - gudgeons [=Gobiones, Gobiobotinae, Armatogobionina, Sarcochilichthyna, Pseudogobioninae] GENUS Abbottina Jordan & Fowler, 1903 - gudgeons, abbottinas [=Pseudogobiops] Species Abbottina binhi Nguyen, in Nguyen & Ngo, 2001 - Cao Bang abbottina Species Abbottina liaoningensis Qin, in Lui & Qin et al., 1987 - Yingkou abbottina Species Abbottina obtusirostris (Wu & Wang, 1931) - Chengtu abbottina Species Abbottina rivularis (Basilewsky, 1855) - North Chinese abbottina [=lalinensis, psegma, sinensis] GENUS Acanthogobio Herzenstein, 1892 - gudgeons Species Acanthogobio guentheri Herzenstein, 1892 - Sinin gudgeon GENUS Belligobio Jordan & Hubbs, 1925 - gudgeons [=Hemibarboides] Species Belligobio nummifer (Boulenger, 1901) - Ningpo gudgeon [=tientaiensis] Species Belligobio pengxianensis Luo et al., 1977 - Sichuan gudgeon GENUS Biwia Jordan & Fowler, 1903 - gudgeons, biwas Species Biwia springeri (Banarescu & Nalbant, 1973) - Springer's gudgeon Species Biwia tama Oshima, 1957 - tama gudgeon Species Biwia yodoensis Kawase & Hosoya, 2010 - Yodo gudgeon Species Biwia zezera (Ishikawa, 1895) - Biwa gudgeon GENUS Coreius Jordan & Starks, 1905 - gudgeons [=Coripareius] Species Coreius cetopsis (Kner, 1867) - cetopsis gudgeon Species Coreius guichenoti (Sauvage & Dabry de Thiersant, 1874) - largemouth bronze gudgeon [=platygnathus, zeni] Species Coreius heterodon (Bleeker, 1865) - bronze gudgeon [=rathbuni, styani] Species Coreius septentrionalis (Nichols, 1925) - Chinese bronze gudgeon [=longibarbus] GENUS Coreoleuciscus -

ML-DSP: Machine Learning with Digital Signal Processing for Ultrafast, Accurate, and Scalable Genome Classification at All Taxonomic Levels Gurjit S
Randhawa et al. BMC Genomics (2019) 20:267 https://doi.org/10.1186/s12864-019-5571-y SOFTWARE ARTICLE Open Access ML-DSP: Machine Learning with Digital Signal Processing for ultrafast, accurate, and scalable genome classification at all taxonomic levels Gurjit S. Randhawa1* , Kathleen A. Hill2 and Lila Kari3 Abstract Background: Although software tools abound for the comparison, analysis, identification, and classification of genomic sequences, taxonomic classification remains challenging due to the magnitude of the datasets and the intrinsic problems associated with classification. The need exists for an approach and software tool that addresses the limitations of existing alignment-based methods, as well as the challenges of recently proposed alignment-free methods. Results: We propose a novel combination of supervised Machine Learning with Digital Signal Processing, resulting in ML-DSP: an alignment-free software tool for ultrafast, accurate, and scalable genome classification at all taxonomic levels. We test ML-DSP by classifying 7396 full mitochondrial genomes at various taxonomic levels, from kingdom to genus, with an average classification accuracy of > 97%. A quantitative comparison with state-of-the-art classification software tools is performed, on two small benchmark datasets and one large 4322 vertebrate mtDNA genomes dataset. Our results show that ML-DSP overwhelmingly outperforms the alignment-based software MEGA7 (alignment with MUSCLE or CLUSTALW) in terms of processing time, while having comparable classification accuracies for small datasets and superior accuracies for the large dataset. Compared with the alignment-free software FFP (Feature Frequency Profile), ML-DSP has significantly better classification accuracy, and is overall faster. We also provide preliminary experiments indicating the potential of ML-DSP to be used for other datasets, by classifying 4271 complete dengue virus genomes into subtypes with 100% accuracy, and 4,710 bacterial genomes into phyla with 95.5% accuracy. -
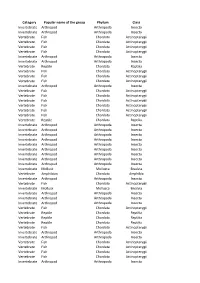
Category Popular Name of the Group Phylum Class Invertebrate
Category Popular name of the group Phylum Class Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate Reptile Chordata Reptilia Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Reptile Chordata Reptilia Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Mollusk Mollusca Bivalvia Vertebrate Amphibian Chordata Amphibia Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Invertebrate Mollusk Mollusca Bivalvia Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate -

Viet Nam Ramsar Information Sheet Published on 16 October 2018
RIS for Site no. 2360, Van Long Wetland Nature Reserve, Viet Nam Ramsar Information Sheet Published on 16 October 2018 Viet Nam Van Long Wetland Nature Reserve Designation date 10 February 2017 Site number 2360 Coordinates 20°23'35"N 105°51'10"E Area 2 736,00 ha https://rsis.ramsar.org/ris/2360 Created by RSIS V.1.6 on - 16 October 2018 RIS for Site no. 2360, Van Long Wetland Nature Reserve, Viet Nam Color codes Fields back-shaded in light blue relate to data and information required only for RIS updates. Note that some fields concerning aspects of Part 3, the Ecological Character Description of the RIS (tinted in purple), are not expected to be completed as part of a standard RIS, but are included for completeness so as to provide the requested consistency between the RIS and the format of a ‘full’ Ecological Character Description, as adopted in Resolution X.15 (2008). If a Contracting Party does have information available that is relevant to these fields (for example from a national format Ecological Character Description) it may, if it wishes to, include information in these additional fields. 1 - Summary Summary Van Long Wetland Nature Reserve is a wetland comprised of rivers and a shallow lake with large amounts of submerged vegetation. The wetland area is centred on a block of limestone karst that rises abruptly from the flat coastal plain of the northern Vietnam. It is located within the Gia Vien district of Ninh Binh Province. The wetland is one of the rarest intact lowland inland wetlands remaining in the Red River Delta, Vietnam. -

Comparative Phylogeography of Two Codistributed Endemic Cyprinids in Southeastern Taiwan
Biochemical Systematics and Ecology 70 (2017) 283e290 Contents lists available at ScienceDirect Biochemical Systematics and Ecology journal homepage: www.elsevier.com/locate/biochemsyseco Comparative phylogeography of two codistributed endemic cyprinids in southeastern Taiwan Tzen-Yuh Chiang a, 1, Yi-Yen Chen a, 1, Teh-Wang Lee b, Kui-Ching Hsu c, * Feng-Jiau Lin d, Wei-Kuang Wang e, Hung-Du Lin f, a Department of Life Sciences, Cheng Kung University, Tainan 701, Taiwan b Taiwan Endemic Species Research Institute, ChiChi, Nantou 552, Taiwan c Department of Industrial Management, National Taiwan University of Science and Technology, Taipei 10607, Taiwan d Tainan Hydraulics Laboratory, National Cheng Kung University, Tainan 709, Taiwan e Department of Environmental Engineering and Science, Feng Chia University, Taichung 407, Taiwan f The Affiliated School of National Tainan First Senior High School, Tainan 701, Taiwan article info abstract Article history: Two cyprinid fishes, Spinibarbus hollandi and Onychostoma alticorpus, are endemic to Received 12 October 2016 southeastern Taiwan. This study examined the phylogeography of these two codistributed Received in revised form 3 December 2016 primary freshwater fishes using mitochondrial DNA cytochrome b sequences (1140 bp) to Accepted 18 December 2016 search for general patterns in the effect of historical changes in southeastern Taiwan. In total, 135 specimens belonging to these two species were collected from five populations. These two codistributed species revealed similar genetic variation patterns. The genetic Keywords: variation in both species was very low, and the geographical distribution of the genetic Spinibarbus hollandi Onychostoma alticorpus variation corresponded neither to the drainage structure nor to the geographical distances Mitochondrial between the samples. -

Two New Species of Shovel-Jaw Carp Onychostoma (Teleostei: Cyprinidae) from Southern Vietnam
Zootaxa 3962 (1): 123–138 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2015 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3962.1.6 http://zoobank.org/urn:lsid:zoobank.org:pub:5872B302-2C4E-4102-B6EC-BC679B3645D4 Two new species of shovel-jaw carp Onychostoma (Teleostei: Cyprinidae) from southern Vietnam HUY DUC HOANG1, HUNG MANH PHAM1 & NGAN TRONG TRAN1 1Viet Nam National University Ho Chi Minh city-University of Science, Faculty of Biology, 227 Nguyen Van Cu, District 5, Ho Chi Minh City, Vietnam. E-mail: [email protected] Corresponding author: Huy D. Hoang: [email protected] Abstract Two new species of large shovel-jaw carps in the genus Onychostoma are described from the upper Krong No and middle Dong Nai drainages of the Langbiang Plateau in southern Vietnam. These new species are known from streams in montane mixed pine and evergreen forests between 140 and 1112 m. Their populations are isolated in the headwaters of the upper Sre Pok River of the Mekong basin and in the middle of the Dong Nai basin. Both species are differentiated from their congeners by a combination of the following characters: transverse mouth opening width greater than head width, 14−17 predorsal scales, caudal-peduncle length 3.9−4.2 times in SL, no barbels in adults and juveniles, a strong serrated last sim- ple ray of the dorsal fin, and small eye diameter (20.3−21.5% HL). Onychostoma krongnoensis sp. nov. is differentiated from Onychostoma dongnaiensis sp. nov. -

Korean Red List of Threatened Species Korean Red List Second Edition of Threatened Species Second Edition Korean Red List of Threatened Species Second Edition
Korean Red List Government Publications Registration Number : 11-1480592-000718-01 of Threatened Species Korean Red List of Threatened Species Korean Red List Second Edition of Threatened Species Second Edition Korean Red List of Threatened Species Second Edition 2014 NIBR National Institute of Biological Resources Publisher : National Institute of Biological Resources Editor in President : Sang-Bae Kim Edited by : Min-Hwan Suh, Byoung-Yoon Lee, Seung Tae Kim, Chan-Ho Park, Hyun-Kyoung Oh, Hee-Young Kim, Joon-Ho Lee, Sue Yeon Lee Copyright @ National Institute of Biological Resources, 2014. All rights reserved, First published August 2014 Printed by Jisungsa Government Publications Registration Number : 11-1480592-000718-01 ISBN Number : 9788968111037 93400 Korean Red List of Threatened Species Second Edition 2014 Regional Red List Committee in Korea Co-chair of the Committee Dr. Suh, Young Bae, Seoul National University Dr. Kim, Yong Jin, National Institute of Biological Resources Members of the Committee Dr. Bae, Yeon Jae, Korea University Dr. Bang, In-Chul, Soonchunhyang University Dr. Chae, Byung Soo, National Park Research Institute Dr. Cho, Sam-Rae, Kongju National University Dr. Cho, Young Bok, National History Museum of Hannam University Dr. Choi, Kee-Ryong, University of Ulsan Dr. Choi, Kwang Sik, Jeju National University Dr. Choi, Sei-Woong, Mokpo National University Dr. Choi, Young Gun, Yeongwol Cave Eco-Museum Ms. Chung, Sun Hwa, Ministry of Environment Dr. Hahn, Sang-Hun, National Institute of Biological Resourses Dr. Han, Ho-Yeon, Yonsei University Dr. Kim, Hyung Seop, Gangneung-Wonju National University Dr. Kim, Jong-Bum, Korea-PacificAmphibians-Reptiles Institute Dr. Kim, Seung-Tae, Seoul National University Dr. -
![Download from the Three [17]](https://docslib.b-cdn.net/cover/9564/download-from-the-three-17-3819564.webp)
Download from the Three [17]
Chou et al. BMC Genomics 2018, 19(Suppl 2):103 DOI 10.1186/s12864-018-4463-x RESEARCH Open Access The aquatic animals’ transcriptome resource for comparative functional analysis Chih-Hung Chou1,2†, Hsi-Yuan Huang1,2†, Wei-Chih Huang1,2†, Sheng-Da Hsu1, Chung-Der Hsiao3, Chia-Yu Liu1, Yu-Hung Chen1, Yu-Chen Liu1,2, Wei-Yun Huang2, Meng-Lin Lee2, Yi-Chang Chen4 and Hsien-Da Huang1,2* From The Sixteenth Asia Pacific Bioinformatics Conference 2018 Yokohama, Japan. 15-17 January 2018 Abstract Background: Aquatic animals have great economic and ecological importance. Among them, non-model organisms have been studied regarding eco-toxicity, stress biology, and environmental adaptation. Due to recent advances in next-generation sequencing techniques, large amounts of RNA-seq data for aquatic animals are publicly available. However, currently there is no comprehensive resource exist for the analysis, unification, and integration of these datasets. This study utilizes computational approaches to build a new resource of transcriptomic maps for aquatic animals. This aquatic animal transcriptome map database dbATM provides de novo assembly of transcriptome, gene annotation and comparative analysis of more than twenty aquatic organisms without draft genome. Results: To improve the assembly quality, three computational tools (Trinity, Oases and SOAPdenovo-Trans) were employed to enhance individual transcriptome assembly, and CAP3 and CD-HIT-EST software were then used to merge these three assembled transcriptomes. In addition, functional annotation analysis provides valuable clues to gene characteristics, including full-length transcript coding regions, conserved domains, gene ontology and KEGG pathways. Furthermore, all aquatic animal genes are essential for comparative genomics tasks such as constructing homologous gene groups and blast databases and phylogenetic analysis. -

(Teleostei: Cyprinidae) from the Middle Chang-Jiang (Yangtze River) Basin, Hubei Province, South China
Zootaxa 3586: 211–221 (2012) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2012 · Magnolia Press Article ISSN 1175-5334 (online edition) urn:lsid:zoobank.org:pub:9835CDA0-A041-4BC9-BAA0-EB320F269790 Microphysogobio nudiventris, a new species of gudgeon (Teleostei: Cyprinidae) from the middle Chang-Jiang (Yangtze River) basin, Hubei Province, South China ZHONG-GUAN JIANG1,2, ER-HU GAO3 & E ZHANG1,4 1Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan 430072, Hubei Province, P. R. China 2Graduate School of Chinese Academy of Sciences, Beijing, 100039, P.R. China 3Academy of Forest Inventory and Planning, State Forestry Administration, 100714, Beijing, P.R. China 4Corresponding author. E-mail: [email protected] Abstract Microphysogobio nudiventris, new species, is described from the Du-He, a tributary flowing into the Han-Jiang of the middle Chang-Jiang (Yangtze River) basin, in Zhushan County, Hubei Province, South China. It belongs in the incompletely scaled group of this genus, but differs from all other species of this group except M. yaluensis, M. rapidus, and M. wulonghensis in the presence of a scaleless midventral region of the body extending more than two-thirds of the distance from the pectoral-fin insertion to the pelvic-fin insertion. This new species differs from M. yaluensis in the slightly concave or straight distal edge of the dorsal fin, interorbital width, and snout length; from M. rapidus in the number of perforated scales on the lateral line and number of pectoral-fin rays, and the placement of the anus; and from M. wulonghensis in having the two lateral lobes of the lower lip posteromedially disconnected, the shape of the median mental pad of the lower lip, and the number of circumpeduncular scales. -

(Cypriniformes: Cyprinidae) from Guangxi Province, Southern China
Zoological Studies 56: 8 (2017) doi:10.6620/ZS.2017.56-08 A New Species of Microphysogobio (Cypriniformes: Cyprinidae) from Guangxi Province, Southern China Shih-Pin Huang1, Yahui Zhao2, I-Shiung Chen3, 4 and Kwang-Tsao Shao5,* 1Biodiversity Research Center, Academia Sinica, Nankang, Taipei 11529, Taiwan. E-mail: [email protected] 2Key Laboratory of Zoological Systematics and Evolution Institute of Zoology, Chinese Academy of Sciences, Chaoyang District, Beijing 100101, China. E-mail: [email protected] 3Institute of Marine Biology, National Taiwan Ocean University, Jhongjheng, Keelung 20224, Taiwan. E-mail: [email protected] 4National Museum of Marine Science and Technology, Jhongjheng, Keelung 20248, Taiwan 5National Taiwan Ocean University, Jhongjheng, Keelung 20224, Taiwan. E-mail: [email protected] (Received 6 January 2017; Accepted 29 March 2017; Published 21 April 2017; Communicated by Benny K.K. Chan) Shih-Pin Huang, Yahui Zhao, I-Shiung Chen, and Kwang-Tsao Shao (2017) Microphysogobio zhangi n. sp., a new cyprinid species is described from Guangxi Province, China. Morphological and molecular evidence based on mitochondrial DNA Cytochrome b (Cyt b) sequence were used for comparing this new species and other related species. The phylogenetic tree topology revealed that this new species is closely related to M. elongatus and M. fukiensis. We also observed the existence of a peculiar trans-river gene flow in the Pearl River and the Yangtze River populations, and speculated that it was caused by an ancient artificial canal, the Lingqu Canal, which forming a pathway directly connecting these two rivers. Key words: Taxonomy, Gudgeon, Cytochrome b, Freshwater Fish, Pearl River. -

Lipid M Etabolism in the Brazilian Teleost Tam Baqui
Colossoma Colossoma ) 20 DOUTORAMENTO EM CIÊNCIA ANIMALDOUTORAMENTO EM CIÊNCIA E EMGENÉTICA MELHORAMENTO ESPECIALIDADE From genes to nutrition: lipid metabolism in in metabolism lipid nutrition: to genes From the Brazilian teleost tambaqui ( macropomum Ferraz Renato Barbosa Maria João Maria Peixoto 20 D Renato Barbosa Ferraz D.ICBAS 2020 From genes to nutrition: lipid metabolism in the Brazilian teleost tambaqui (Colossoma macropomum) From genes to nutrition: lipid metabolism in the Brazilian teleost tambaqui (Colossoma macropomum) Renato Barbosa Ferraz INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR Renato Barbosa Ferraz 2020 From genes to nutrition: lipid metabolism in the Brazilian teleost tambaqui (Colossoma macropomum) Tese de Candidatura ao grau de Doutor em Ciências Animal - Especialidade em Genética e Melhoramento - submetida ao Instituto de Ciências Biomédicas Abel Salazar da Universidade do Porto. Orientador Professor Doutor Luís Filipe Castro, Professor da Faculdade de Ciências da Universidade do Porto. Co-orientador Doutor Rodrigo Ozório, Investigador no CIIMAR. Co-orientador Professora Doutora Ana Lúcia Salário, Professora da Universidade Federal de Viçosa (Brasil). Financial support This study was supported by CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico – Brazil, for the doctoral scholarship to student Renato Barbosa Ferraz with process number 201864 / 2015-0. And, by COMPETE 2020, Portugal 2020 and the European Union through the ERDF, grant number 031342, and by FCT through national funds (PTDC/CTA-AMB/31342/2017) and the strategic project UID/Multi/04423/2019 granted to CIIMAR. The research reported in this thesis was conducted at: 1) Instituto de Ciências Biomédicas Abel Salazar (ICBAS), Universidade do Porto (Portugal), 2) Centro Interdisciplinar de Investigação Marinha e Ambiental (CIIMAR), Universidade do Porto and 3) Departamento de Biologia Animal, Universidade Federal de Viçosa (Brasil).