Rubber Elasticity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

10-1 CHAPTER 10 DEFORMATION 10.1 Stress-Strain Diagrams And
EN380 Naval Materials Science and Engineering Course Notes, U.S. Naval Academy CHAPTER 10 DEFORMATION 10.1 Stress-Strain Diagrams and Material Behavior 10.2 Material Characteristics 10.3 Elastic-Plastic Response of Metals 10.4 True stress and strain measures 10.5 Yielding of a Ductile Metal under a General Stress State - Mises Yield Condition. 10.6 Maximum shear stress condition 10.7 Creep Consider the bar in figure 1 subjected to a simple tension loading F. Figure 1: Bar in Tension Engineering Stress () is the quotient of load (F) and area (A). The units of stress are normally pounds per square inch (psi). = F A where: is the stress (psi) F is the force that is loading the object (lb) A is the cross sectional area of the object (in2) When stress is applied to a material, the material will deform. Elongation is defined as the difference between loaded and unloaded length ∆푙 = L - Lo where: ∆푙 is the elongation (ft) L is the loaded length of the cable (ft) Lo is the unloaded (original) length of the cable (ft) 10-1 EN380 Naval Materials Science and Engineering Course Notes, U.S. Naval Academy Strain is the concept used to compare the elongation of a material to its original, undeformed length. Strain () is the quotient of elongation (e) and original length (L0). Engineering Strain has no units but is often given the units of in/in or ft/ft. ∆푙 휀 = 퐿 where: is the strain in the cable (ft/ft) ∆푙 is the elongation (ft) Lo is the unloaded (original) length of the cable (ft) Example Find the strain in a 75 foot cable experiencing an elongation of one inch. -
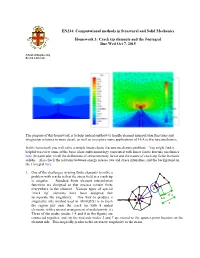
Crack Tip Elements and the J Integral
EN234: Computational methods in Structural and Solid Mechanics Homework 3: Crack tip elements and the J-integral Due Wed Oct 7, 2015 School of Engineering Brown University The purpose of this homework is to help understand how to handle element interpolation functions and integration schemes in more detail, as well as to explore some applications of FEA to fracture mechanics. In this homework you will solve a simple linear elastic fracture mechanics problem. You might find it helpful to review some of the basic ideas and terminology associated with linear elastic fracture mechanics here (in particular, recall the definitions of stress intensity factor and the nature of crack-tip fields in elastic solids). Also check the relations between energy release rate and stress intensities, and the background on the J integral here. 1. One of the challenges in using finite elements to solve a problem with cracks is that the stress field at a crack tip is singular. Standard finite element interpolation functions are designed so that stresses remain finite a everywhere in the element. Various types of special b c ‘crack tip’ elements have been designed that 3L/4 incorporate the singularity. One way to produce a L/4 singularity (the method used in ABAQUS) is to mesh L the region just near the crack tip with 8 noded elements, with a special arrangement of nodal points: (i) Three of the nodes (nodes 1,4 and 8 in the figure) are connected together, and (ii) the mid-side nodes 2 and 7 are moved to the quarter-point location on the element side. -

Engineering Viscoelasticity
ENGINEERING VISCOELASTICITY David Roylance Department of Materials Science and Engineering Massachusetts Institute of Technology Cambridge, MA 02139 October 24, 2001 1 Introduction This document is intended to outline an important aspect of the mechanical response of polymers and polymer-matrix composites: the field of linear viscoelasticity. The topics included here are aimed at providing an instructional introduction to this large and elegant subject, and should not be taken as a thorough or comprehensive treatment. The references appearing either as footnotes to the text or listed separately at the end of the notes should be consulted for more thorough coverage. Viscoelastic response is often used as a probe in polymer science, since it is sensitive to the material’s chemistry and microstructure. The concepts and techniques presented here are important for this purpose, but the principal objective of this document is to demonstrate how linear viscoelasticity can be incorporated into the general theory of mechanics of materials, so that structures containing viscoelastic components can be designed and analyzed. While not all polymers are viscoelastic to any important practical extent, and even fewer are linearly viscoelastic1, this theory provides a usable engineering approximation for many applications in polymer and composites engineering. Even in instances requiring more elaborate treatments, the linear viscoelastic theory is a useful starting point. 2 Molecular Mechanisms When subjected to an applied stress, polymers may deform by either or both of two fundamen- tally different atomistic mechanisms. The lengths and angles of the chemical bonds connecting the atoms may distort, moving the atoms to new positions of greater internal energy. -

Viscoelasticity of Filled Elastomers: Determination of Surface-Immobilized Components and Their Role in the Reinforcement of SBR-Silica Nanocomposites
Viscoelasticity of Filled Elastomers: Determination of Surface-Immobilized Components and their Role in the Reinforcement of SBR-Silica Nanocomposites Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) genehmigt durch die Naturwissenschaftliche Fakultät II Institut für Physik der Martin-Luther-Universität Halle-Wittenberg vorgelegt von M. Sc. Anas Mujtaba geboren am 03.04.1980 in Lahore, Pakistan Halle (Saale), January 15th, 2014 Gutachter: 1. Prof. Dr. Thomas Thurn-Albrecht 2. Prof. Dr. Manfred Klüppel 3. Prof. Dr. Rene Androsch Öffentliche Verteidigung: July 3rd, 2014 In loving memory of my beloved Sister Rabbia “She will be in my Heart ξ by my Side for the Rest of my Life” Contents 1 Introduction 1 2 Theoretical Background 5 2.1 Elastomers . .5 2.1.1 Fundamental Theories on Rubber Elasticity . .7 2.2 Fillers . 10 2.2.1 Carbon Black . 12 2.2.2 Silica . 12 2.3 Filled Rubber Reinforcement . 15 2.3.1 Occluded Rubber . 16 2.3.2 Payne Effect . 17 2.3.3 The Kraus Model for the Strain-Softening Effect . 18 2.3.4 Filler Network Reinforcement . 20 3 Experimental Methods 29 3.1 Dynamic Mechanical Analysis . 29 3.1.1 Temperature-dependent Measurement (Temperature Sweeps) . 31 3.1.2 Time-Temperature Superposition (Master Curves) . 31 3.1.3 Strain-dependent Measurement (Payne Effect) . 33 3.2 Low-field NMR . 34 3.2.1 Theoretical Concept . 34 3.2.2 Experimental Details . 39 4 Optimizing the Tire Tread 43 4.1 Relation Between Friction and the Mechanical Properties of Tire Rubbers 46 4.2 Usage of tan δ As Loss Parameter . -

Elasticity and Viscoelasticity
CHAPTER 2 Elasticity and Viscoelasticity CHAPTER 2.1 Introduction to Elasticity and Viscoelasticity JEAN LEMAITRE Universite! Paris 6, LMT-Cachan, 61 avenue du President! Wilson, 94235 Cachan Cedex, France For all solid materials there is a domain in stress space in which strains are reversible due to small relative movements of atoms. For many materials like metals, ceramics, concrete, wood and polymers, in a small range of strains, the hypotheses of isotropy and linearity are good enough for many engineering purposes. Then the classical Hooke’s law of elasticity applies. It can be de- rived from a quadratic form of the state potential, depending on two parameters characteristics of each material: the Young’s modulus E and the Poisson’s ratio n. 1 c * ¼ A s s ð1Þ 2r ijklðE;nÞ ij kl @c * 1 þ n n eij ¼ r ¼ sij À skkdij ð2Þ @sij E E Eandn are identified from tensile tests either in statics or dynamics. A great deal of accuracy is needed in the measurement of the longitudinal and transverse strains (de Æ10À6 in absolute value). When structural calculations are performed under the approximation of plane stress (thin sheets) or plane strain (thick sheets), it is convenient to write these conditions in the constitutive equation. Plane stress ðs33 ¼ s13 ¼ s23 ¼ 0Þ: 2 3 1 n 6 À 0 7 2 3 6 E E 72 3 6 7 e11 6 7 s11 6 7 6 1 76 7 4 e22 5 ¼ 6 0 74 s22 5 ð3Þ 6 E 7 6 7 e12 4 5 s12 1 þ n Sym E Handbook of Materials Behavior Models Copyright # 2001 by Academic Press. -

A Bayesian Surrogate Constitutive Model to Estimate Failure Probability of Rubber-Like Materials
A Bayesian Surrogate Constitutive Model to Estimate Failure Probability of Rubber-Like Materials Aref Ghaderia,b, Vahid Morovatia,c, Roozbeh Dargazany1a,d aDepartment of Civil and Environmental Engineering, Michigan State University [email protected] [email protected] [email protected] Abstract In this study, a stochastic constitutive modeling approach for elastomeric materials is developed to consider uncer- tainty in material behavior and its prediction. This effort leads to a demonstration of the deterministic approaches error compared to probabilistic approaches in order to calculate the probability of failure. First, the Bayesian lin- ear regression model calibration approach is employed for the Carroll model representing a hyperelastic constitutive model. The developed model is calibrated based on the Maximum Likelihood Estimation (MLE) and Maximum a Priori (MAP) estimation. Next, a Gaussian process (GP) as a non-parametric approach is utilized to estimate the probabilistic behavior of elastomeric materials. In this approach, hyper-parameters of the radial basis kernel in GP are calculated using L-BFGS method. To demonstrate model calibration and uncertainty propagation, these approaches are conducted on two experimental data sets for silicon-based and polyurethane-based adhesives, with four samples from each material. These uncertainties stem from model, measurement, to name but a few. Finally, failure proba- bility calculation analysis is conducted with First Order Reliability Method (FORM) analysis and Crude Monte Carlo (CMC) simulation for these data sets by creating a limit state function based on the stochastic constitutive model at failure stretch. Furthermore, sensitivity analysis is used to show the importance of each parameter of the probability of failure. -

The Thermodynamic Properties of Elastomers: Equation of State and Molecular Structure
CH 351L Wet Lab 4 / p. 1 The Thermodynamic Properties of Elastomers: Equation of State and Molecular Structure Objective To determine the macroscopic thermodynamic equation of state of an elastomer, and relate it to its microscopic molecular properties. Introduction We are all familiar with the very useful properties of such objects as rubber bands, solid rubber balls, and tires. Materials such as these, which are capable of undergoing large reversible extensions and compressions, are called elastomers. An example of such a material is natural rubber, obtained from the plant Hevea brasiliensis. An elastomer has rather unusual physical properties; for example, an ordinary elastic band can be stretched up to 15 times its original length and then be restored to its original size. Although we might initially consider elastomers to be a solids, many have isothermal compressibilities comparable with that of liquids (e.g. toluene), about 10-4 atm-1 (compared with solids such as polystyrene or aluminum which have values of ~10–6 atm–1). Certain evidence suggests that an elastomer is a disordered "solid," i.e., a glass, that cannot flow as a result of internal, structural restrictions. The reversible deformability of an elastomer is reminiscent of a gas. In fact the term elastic was first used by Robert Boyle (1660) in describing a gas, "There is a spring or elastical power in the air in which we live." In this experiment, you will encounter certain formal thermodynamic similarities between an elastomer and a gas. One of the rather dramatic and anomalous properties of an elastomer is that once brought to an extended form, it contracts upon heating. -

Cross-Linked Polymers and Rubber Elasticity Chapter 9 (Sperling)
Cross-linked Polymers and Rubber Elasticity Chapter 9 (Sperling) • Definition of Rubber Elasticity and Requirements • Cross-links, Networks, Classes of Elastomers (sections 1-3, 16) • Simple Theory of Rubber Elasticity (sections 4-8) – Entropic Origin of Elastic Retractive Forces – The Ideal Rubber Behavior • Departures from the Ideal Rubber Behavior (sections 9-11) – Non-zero Energy Contribution to the Elastic Retractive Forces – Stress-induced Crystallization and Limited Extensibility of Chains (How to make better elastomers: High Strength and High Modulus) – Network Defects (dangling chains, loops, trapped entanglements, etc..) – Semi-empirical Mooney-Rivlin Treatment (Affine vs Non-Affine Deformation) Definition of Rubber Elasticity and Requirements • Definition of Rubber Elasticity: Very large deformability with complete recoverability. • Molecular Requirements: – Material must consist of polymer chains. Need to change conformation and extension under stress. – Polymer chains must be highly flexible. Need to access conformational changes (not w/ glassy, crystalline, stiff mat.) – Polymer chains must be joined in a network structure. Need to avoid irreversible chain slippage (permanent strain). One out of 100 monomers must connect two different chains. Connections (covalent bond, crystallite, glassy domain in block copolymer) Cross-links, Networks and Classes of Elastomers • Chemical Cross-linking Process: Sol-Gel or Percolation Transition • Gel Characteristics: – Infinite Viscosity – Non-zero Modulus – One giant Molecule – Solid -

Chapter 10: Elasticity and Oscillations
Chapter 10 Lecture Outline 1 Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Chapter 10: Elasticity and Oscillations •Elastic Deformations •Hooke’s Law •Stress and Strain •Shear Deformations •Volume Deformations •Simple Harmonic Motion •The Pendulum •Damped Oscillations, Forced Oscillations, and Resonance 2 §10.1 Elastic Deformation of Solids A deformation is the change in size or shape of an object. An elastic object is one that returns to its original size and shape after contact forces have been removed. If the forces acting on the object are too large, the object can be permanently distorted. 3 §10.2 Hooke’s Law F F Apply a force to both ends of a long wire. These forces will stretch the wire from length L to L+L. 4 Define: L The fractional strain L change in length F Force per unit cross- stress A sectional area 5 Hooke’s Law (Fx) can be written in terms of stress and strain (stress strain). F L Y A L YA The spring constant k is now k L Y is called Young’s modulus and is a measure of an object’s stiffness. Hooke’s Law holds for an object to a point called the proportional limit. 6 Example (text problem 10.1): A steel beam is placed vertically in the basement of a building to keep the floor above from sagging. The load on the beam is 5.8104 N and the length of the beam is 2.5 m, and the cross-sectional area of the beam is 7.5103 m2. -

Elastomeric Materials
ELASTOMERIC MATERIALS TAMPERE UNIVERSITY OF TECHNOLOGY THE LABORATORY OF PLASTICS AND ELASTOMER TECHNOLOGY Kalle Hanhi, Minna Poikelispää, Hanna-Mari Tirilä Summary On this course the students will get the basic information on different grades of rubber and thermoelasts. The chapters focus on the following subjects: - Introduction - Rubber types - Rubber blends - Thermoplastic elastomers - Processing - Design of elastomeric products - Recycling and reuse of elastomeric materials The first chapter introduces shortly the history of rubbers. In addition, it cover definitions, manufacturing of rubbers and general properties of elastomers. In this chapter students get grounds to continue the studying. The second chapter focus on different grades of elastomers. It describes the structure, properties and application of the most common used rubbers. Some special rubbers are also covered. The most important rubber type is natural rubber; other generally used rubbers are polyisoprene rubber, which is synthetic version of NR, and styrene-butadiene rubber, which is the most important sort of synthetic rubber. Rubbers always contain some additives. The following chapter introduces the additives used in rubbers and some common receipts of rubber. The important chapter is Thermoplastic elastomers. Thermoplastic elastomers are a polymer group whose main properties are elasticity and easy processability. This chapter introduces the groups of thermoplastic elastomers and their properties. It also compares the properties of different thermoplastic elastomers. The chapter Processing give a short survey to a processing of rubbers and thermoplastic elastomers. The following chapter covers design of elastomeric products. It gives the most important criteria in choosing an elastomer. In addition, dimensioning and shaping of elastomeric product are discussed The last chapter Recycling and reuse of elastomeric materials introduces recycling methods. -
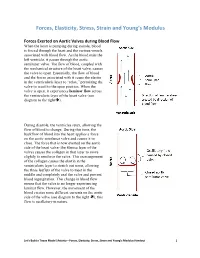
Forces, Elasticity, Stress, Strain and Young's Modulus Handout
Forces, Elasticity, Stress, Strain and Young’s Modulus Forces Exerted on Aortic Valves during Blood Flow When the heart is pumping during systole, blood is forced through the heart and the various vessels associated with blood flow. As the blood exits the left ventricle, it passes through the aortic semilunar valve. The flow of blood, coupled with the mechanical structure of the heart valve, causes the valve to open. Essentially, the flow of blood and the forces associated with it cause the elastin in the ventricularis layer to “relax,” permitting the valve to recoil to the open position. When the valve is open, it experiences laminar flow across the ventricularis layer of the heart valve (see diagram to the right). During diastole, the ventricles relax, allowing the flow of blood to change. During this time, the backflow of blood into the heart applies a force on the aortic semilunar valve and causes it to close. The force that is now exerted on the aortic side of the heart valve (the fibrosa layer of the valve) causes the collagen in that layer to move slightly to reinforce the valve. This rearrangement of the collagen causes the elastin in the ventricularis layer to stretch out some, allowing the three leaflets of the valve to meet in the middle and completely seal the valve and prevent blood regurgitation. This change in blood flow means that the valve is no longer experiencing laminar flow. However, the movement of the blood creates some different currents on the aortic side of the valve (see diagram to the right ); this flow is oscillatory in nature. -

Rubber As an Aid to Teach Thermodynamics∗ the Discovery by a Blind Scientist
GENERAL ARTICLE Rubber as an Aid to Teach Thermodynamics∗ The Discovery by a Blind Scientist Geethamma V G and Sampath V The behaviour of rubber differs from that of conventional materials. Rubber heats up on stretching and cools on re- traction. Also, stretched rubber shrinks on heating (ther- moelastic shrinkage) while a stretched metal elongates. The elastic recovery of rubber is due to its tendency to maximize the entropy. The same property also causes the thermoe- lastic shrinkage. Metallic materials possess energy elastic- Geethamma V G was a ity, while ideal rubber possesses entropy elasticity. The ther- Fulbright Fellow at modynamic behaviour of rubber is similar to that of gaseous University of Illinois, USA materials. Hence rubber can be used as an aid for teaching and a Royal Society International Postdoctoral thermodynamics. Fellow at Cavendish Lab, University of Cambridge, UK. She was also a Young 1. Gough–Joule Effect Scientist Awardee. John Gough was not born blind. But by a sheer quirk of fate, he lost his eyesight due to smallpox before turning three. However, his senses of touch and hearing were intact, and he was bestowed with a sharp inquisitive mind and adequate skills for experimen- V Sampath is presently a tation. He tended to be philosophical too. John Dalton, a well Professor of Metallurgical known British scientist, who proposed the atomic theory, helped and Materials Engg. at IIT Gough by reading out to and writing for him. He had great admi- Madras. He holds a PhD from IISc, Bengaluru and has ration for Gough. three decades of research and In 1805, Gough observed two important properties of rubber [1].