Small-Molecule Inhibition of TLR8 Through Stabilization of Its Resting State
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TLR8-Dependent Immune Responses Stimulating Sequence-Specific
Identification of RNA Sequence Motifs Stimulating Sequence-Specific TLR8-Dependent Immune Responses This information is current as Alexandra Forsbach, Jean-Guy Nemorin, Carmen Montino, of September 28, 2021. Christian Müller, Ulrike Samulowitz, Alain P. Vicari, Marion Jurk, George K. Mutwiri, Arthur M. Krieg, Grayson B. Lipford and Jörg Vollmer J Immunol 2008; 180:3729-3738; ; doi: 10.4049/jimmunol.180.6.3729 Downloaded from http://www.jimmunol.org/content/180/6/3729 References This article cites 55 articles, 25 of which you can access for free at: http://www.jimmunol.org/content/180/6/3729.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 28, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Identification of RNA Sequence Motifs Stimulating Sequence-Specific TLR8-Dependent Immune Responses1 Alexandra Forsbach,* Jean-Guy Nemorin,† Carmen Montino,* Christian Mu¨ller,* Ulrike Samulowitz,* Alain P. -

Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods
pharmaceutics Review Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods Nasir Javaid, Farzana Yasmeen and Sangdun Choi * Department of Molecular Science and Technology, Ajou University, Suwon 16499, Korea * Correspondence: [email protected]; Tel.: +82-31-219-2600 Received: 9 July 2019; Accepted: 28 August 2019; Published: 1 September 2019 Abstract: The built-in innate immunity in the human body combats various diseases and their causative agents. One of the components of this system is Toll-like receptors (TLRs), which recognize structurally conserved molecules derived from microbes and/or endogenous molecules. Nonetheless, under certain conditions, these TLRs become hypofunctional or hyperfunctional, thus leading to a disease-like condition because their normal activity is compromised. In this regard, various small-molecule drugs and recombinant therapeutic proteins have been developed to treat the relevant diseases, such as rheumatoid arthritis, psoriatic arthritis, Crohn’s disease, systemic lupus erythematosus, and allergy. Some drugs for these diseases have been clinically approved; however, their efficacy can be enhanced by conventional or targeted drug delivery systems. Certain delivery vehicles such as liposomes, hydrogels, nanoparticles, dendrimers, or cyclodextrins can be employed to enhance the targeted drug delivery. This review summarizes the TLR signaling pathway, associated diseases and their treatments, and the ways to efficiently deliver the drugs to a target site. Keywords: Toll-like receptor; immunological disease; therapeutic; drug delivery method 1. Introduction The immune system in an organism is the protective system combating pathogenic and/or abnormal conditions. Innate and adaptive immune systems are two basic components in vertebrates. Adaptive immunity is mediated by T and B lymphocytes with the help of their antigen-specific receptors that are encoded as a result of hypermutability and rearrangements in a genomic region of the organism. -

Cx3cr1 Mediates the Development of Monocyte-Derived Dendritic Cells During Hepatic Inflammation
CX3CR1 MEDIATES THE DEVELOPMENT OF MONOCYTE-DERIVED DENDRITIC CELLS DURING HEPATIC INFLAMMATION. Supplementary material Supplementary Figure 1: Liver CD45+ myeloid cells were pre-gated for Ly6G negative cells for excluding granulocytes and HDCs subsequently analyzed among the cells that were CD11c+ and had high expression of MHCII. Supplementary Table 1 low/- high + Changes in gene expression between CX3CR1 and CX3CR1 CD11b myeloid hepatic dendritic cells (HDCs) from CCl4-treated mice high Genes up-regulated in CX3CR1 HDCs Gene Fold changes P value Full name App 4,01702 5,89E-05 amyloid beta (A4) precursor protein C1qa 9,75881 1,69E-22 complement component 1, q subcomponent, alpha polypeptide C1qb 9,19882 3,62E-20 complement component 1, q subcomponent, beta polypeptide Ccl12 2,51899 0,011769 chemokine (C-C motif) ligand 12 Ccl2 6,53486 6,37E-11 chemokine (C-C motif) ligand 2 Ccl3 4,99649 5,84E-07 chemokine (C-C motif) ligand 3 Ccl4 4,42552 9,62E-06 chemokine (C-C motif) ligand 4 Ccl6 3,9311 8,46E-05 chemokine (C-C motif) ligand 6 Ccl7 2,60184 0,009272 chemokine (C-C motif) ligand 7 Ccl9 4,17294 3,01E-05 chemokine (C-C motif) ligand 9 Ccr2 3,35195 0,000802 chemokine (C-C motif) receptor 2 Ccr5 3,23358 0,001222 chemokine (C-C motif) receptor 5 Cd14 6,13325 8,61E-10 CD14 antigen Cd36 2,94367 0,003243 CD36 antigen Cd44 4,89958 9,60E-07 CD44 antigen Cd81 6,49623 8,24E-11 CD81 antigen Cd9 3,06253 0,002195 CD9 antigen Cdkn1a 4,65279 3,27E-06 cyclin-dependent kinase inhibitor 1A (P21) Cebpb 6,6083 3,89E-11 CCAAT/enhancer binding protein (C/EBP), -

TLR8 Differences Between Human TLR7 and Synthetic TLR Agonists
Synthetic TLR Agonists Reveal Functional Differences between Human TLR7 and TLR8 Keith B. Gorden, Kevin S. Gorski, Sheila J. Gibson, Ross M. Kedl, William C. Kieper, Xiaohong Qiu, Mark A. Tomai, This information is current as Sefik S. Alkan and John P. Vasilakos of September 29, 2021. J Immunol 2005; 174:1259-1268; ; doi: 10.4049/jimmunol.174.3.1259 http://www.jimmunol.org/content/174/3/1259 Downloaded from References This article cites 36 articles, 14 of which you can access for free at: http://www.jimmunol.org/content/174/3/1259.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 29, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Synthetic TLR Agonists Reveal Functional Differences between Human TLR7 and TLR8 Keith B. Gorden, Kevin S. Gorski, Sheila J. Gibson, Ross M. Kedl,1 William C. -

TLR8 on Dendritic Cells and TLR9 on B Cells Restrain TLR7-Mediated Spontaneous Autoimmunity in C57BL/6 Mice
TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice Benoit Desnuesa,b,c,1, Amanda Beatriz Macedoa,b,c,1, Annie Roussel-Quevala,b,c, Johnny Bonnardela,b,c, Sandrine Henria,b,c, Olivier Demariaa,b,c,2, and Lena Alexopouloua,b,c,3 aCentre d’Immunologie de Marseille-Luminy, Aix-Marseille University UM 2, 13288 Marseille, France; bInstitut National de la Santé et de la Recherche Médicale, Unité Mixte de Recherche 1104, 13288 Marseille, France; and cCentre National de la Recherche Scientifique, Unité Mixte de Recherche 7280, 13288 Marseille, France Edited* by Richard A. Flavell, Yale School of Medicine, Howard Hughes Medical Institute, New Haven, CT, and approved December 18, 2013 (received for review August 5, 2013) Systemic lupus erythematosus (SLE) is a complex autoimmune endosomal localization isolate TLR3, TLR7, TLR8, and disease with diverse clinical presentations characterized by the TLR9 away from self-nucleic acids in the extracellular space, still presence of autoantibodies to nuclear components. Toll-like re- self-RNA or -DNA can become a potent trigger of cell activation ceptor (TLR)7, TLR8, and TLR9 sense microbial or endogenous nucleic when transported into TLR-containing endosomes, and such acids and are implicated in the development of SLE. In mice TLR7- recognition can result in sterile inflammation and autoimmunity, deficiency ameliorates SLE, but TLR8- or TLR9-deficiency exacerbates including SLE (4, 15, 16). The connection of the endosomal the disease because of increased TLR7 response. Thus, both TLR8 TLRs with SLE originates mainly from mouse models, where and TLR9 control TLR7 function, but whether TLR8 and TLR9 act in TLR7 signaling seems to play a central role. -

Human TLR7 Or TLR8 Independently Confer Responsiveness to the Antiviral Compound R-848
CORRESPONDENCE bers of this family (TLR1 to In the quest to identify ligands for TLRs, Human TLR7 or TLR8 TLR10) have been reported to we also screened immunostimulatory synthet- date. TLR2, TLR4 and TLR5 ic compounds for their potential to activate are crucial for the recognition HEK293 cells that were transiently transfect- independently confer of peptidoglycan, lipopolysac- ed with TLR cDNAs and a NF-κB luciferase charide and flagellin, respec- reporter plasmid. We found that R-848 responsiveness to the tively3–5. TLR6 associates with induced NF-κB activation in HEK293 cells TLR2 and recognizes lipopro- transfected with human TLR8 (GenBank antiviral compound teins from mycoplasma6. TLR9 accession number AF245703) in a dose- detects bacterial DNA contain- dependent manner (Fig. 1). In accordance ing unmethylated CpG motifs with the findings of Hemmi et al., human and R-848 and TLR3 activates immune murine TLR7 (GenBank accession numbers cells in response to double- AF240467 and AY035889) also mediate 1, 2, 1 MARION JURK *, FLORIAN HEIL *, JÖRG VOLLMER , stranded RNA7–9. The natural R-848 recognition (Fig. 1)10. TLR7 showed a CHRISTIAN SCHETTER1,ARTHUR M. KRIEG1,HERMANN ligands for TLR1, TLR7, higher sensitivity to R-848, but TLR8 was WAGNER2,GRAYSON LIPFORD1 AND STEFAN BAUER2 TLR8 and TLR10 are not able to induce NF-κB more effectively than known, although a synthetic TLR7 when higher concentrations of R-848 1Coley Pharmaceutical GmbH, Elisabeth-Selbert-Strasse 9, 40764 Langenfeld, Germany and Coley Pharmaceutical Group, Inc., 93 Worcester compound with antiviral activi- were used (Fig. 1). In contrast, HEK293 cells St.,Wellesley, MA 02481, USA. -

A CROSS-SECTIONAL STUDY on the EFFECT of HIV VIRION and BACTERIAL LPS on MEMORY B CELL APOPTOSIS by WEI JIANG Submitted in Part
A CROSS-SECTIONAL STUDY ON THE EFFECT OF HIV VIRION AND BACTERIAL LPS ON MEMORY B CELL APOPTOSIS by WEI JIANG Submitted in partial fulfillment of requirements for the degree of Master of Science Dissertation Advisor: Dr. Daniel Tisch Department of Epidemiology and Biostatistics CASE WESTEN RESERVE UNIVERSITY May, 2012 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Wei Jiang candidate for the Master of Science degree*. (signed) Daniel Tisch (chair of the committee) Daniel Tisch XiaoFeng Zhu Mark D. Schluchter (date) March27th, 2012 *We also certify that written approval has been obtained for any proprietary material contained therein. 2 Dedicated to my dear husband Zhuang Wan whose patience and encouragement have always given me great support 3 I very much appreciate Dr. PingFu Fu for his support and guidance. I would like to acknowledge my advisor Dr. Daniel Tisch, and committee members Dr. Mark D. Schluchter and Dr. XiaoFeng Zhu. This work was supported by STERIS grant at Case Western Reserve University and NIAID R01AI091526. 4 TABLE OF CONTENTS Abstract 9 Introduction 11 Specific aims 20 Study methods 21 Results 36 Discussion 59 References 66 5 LIST OF TABLES Table 1. Characteristics of HIV-negative control and 36 HIV-infected subjects Table 2. Dependent variable and independent variables 37 Table 3. Correlation test in all subjects 46 Table 4. Correlation test in control subjects 46 Table 5. Correlation test in HIV-infected subjects 47 Table 6. Independent variables include plasma level of FasL, 48 Fas geometric mean expression on memory B cells, or HIV status among all subjects in a linear regression model by forward selection Table 7. -
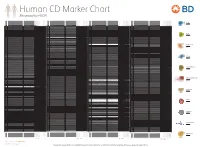
Reviewed by HLDA1
Human CD Marker Chart Reviewed by HLDA1 T Cell Key Markers CD3 CD4 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD8 CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD74 DHLAG, HLADG, Ia-g, li, invariant chain HLA-DR, CD44 + + + + + + CD158g KIR2DS5 + + CD248 TEM1, Endosialin, CD164L1, MGC119478, MGC119479 Collagen I/IV Fibronectin + ST6GAL1, MGC48859, SIAT1, ST6GALL, ST6N, ST6 b-Galactosamide a-2,6-sialyl- CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD75 CD22 CD158h KIR2DS1, p50.1 HLA-C + + CD249 APA, gp160, EAP, ENPEP + + tranferase, Sialo-masked lactosamine, Carbohydrate of a2,6 sialyltransferase + – – + – – + – – CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD75S a2,6 Sialylated lactosamine CD22 (proposed) + + – – + + – + + + CD158i KIR2DS4, p50.3 HLA-C + – + CD252 TNFSF4, -

Final Program
Dear Colleagues, Welcome to the 20th Annual Meeting of the Federation of Clinical Immunology Societies, FOCIS 2020, and the first- ever virtual offering of THE meeting in translational immunology. The FOCIS 2020 Scientific Program is presented by world-renowned leaders from academia and industry presenting new developments in immune therapies, immune diseases, and developing technologies to measure and assess the immune system. Participants will share in best practices for data sharing and assay standardization and the rapid emergence of ever-evolving technologies, and the application of immunological discovery to the challenges facing our global community, that includes the COVID-19 pandemic. This year FOCIS celebrates its 20th anniversary. Since its inception in 2001, FOCIS has continued to strengthen its interdisciplinary approach to clinical immunology and broaden its scope in pursuit of our mission to improve human health through immunology. In our 20th year, FOCIS has 59 Member Societies and 1,000 individual members bridging multiple disciplines with a common focus on immune-mediated disease. The FOCIS Annual Meeting is the leading forum for researchers and clinicians from a multitude of specialties, along with industry and governmental representatives, to come together and share the latest breakthroughs. We value the participation of each of you who have contributed to FOCIS’ success over the past 20 years, and we know that you will continue to empower the efforts of our community to secure the future of the field. We encourage you to get involved in FOCIS. Join FOCIS as a member and join our efforts to promote education, research, and clinical care through translational immunology. -
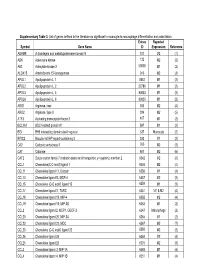
Supplementary Table 3. List of Genes Defined in the Literature As Significant in Monocyte-To-Macrophage Differentiation and Polarization
Supplementary Table 3. List of genes defined in the literature as significant in monocyte-to-macrophage differentiation and polarization. Entrez Reported Symbol Gene Name ID Expression Reference ADAM8 A disintegrin and metalloproteinase domain 8 101 M2 (1) ADK Adenosine kinase 132 M2 (2) AK3 Adenylate kinase 3 50808 M1 (2) ALOX15 Arachidonate 15-lipoxygenase 246 M2 (3) APOL1 Apolipoprotein L, 1 8542 M1 (2) APOL2 Apolipoprotein L, 2 23780 M1 (2) APOL3 Apolipoprotein L, 3 80833 M1 (2) APOL6 Apolipoprotein L, 6 80830 M1 (2) ARG1 Arginase, liver 383 M2 (4) ARG2 Arginase, type II 384 M2 (5) ATF3 Activating transcription factor 3 467 M1 (2) BCL2A1 BCL2-related protein A1 597 M1 (2) BID BH3 interacting domain death agonist 637 Monocyte (2) BIRC3 Baculoviral IAP repeat-containing 3 330 M1 (2) CA2 Carbonic anhydrase II 760 M2 (2) CAT Catalase 847 M2 (6) CAT2 Solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 6542 M2 (6) CCL1 Chemokine(C-C motif) ligand 1 6346 M2 (4) CCL11 Chemokine ligand 11, Eotaxin 6356 M1 (4) CCL13 Chemokine ligand13, MCP-4 6357 M2 (2) CCL15 Chemokine (C-C motif) ligand 15 6359 M1 (2) CCL17 Chemokine ligand17, TARC 6361 M1 & M2 (4) CCL18 Chemokine ligand 18, MIP-4 6362 M2 (4) CCL19 Chemokine ligand 19, MIP-3B 6363 M1 (2) CCL2 Chemokine ligand 2, MCP1, GDCF-2 6347 Macrophage (2) CCL20 Chemokine ligand 20, MIP-3A 6364 M1 (2) CCL22 Chemokine ligand 22, MDC 6367 M2 (7) CCL23 Chemokine (C-C motif) ligand 23 6368 M2 (2) CCL24 Chemokine ligand 24 6369 M2 (4) CCL25 Chemokine ligand 25 6370 M2 (8) CCL3 Chemokine -

Downloaded Using the R Package “TCGA Biolinks” (12)
2872 Original Article Systemic immune microenvironment and regulatory network analysis in patients with lung adenocarcinoma Libao Liu1#, Shilei Xu2#, Lei Huang3, Jinyuan He1, Gang Liu4, Shaohong Ma1, Yimin Weng1, Shaohong Huang1 1Department of Cardiothoracic Surgery, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China; 2Department of General Surgery, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China; 3Department of Nursing, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China; 4Department of Cardiovascular Medicine, First Affiliated Hospital of Sun at-senY University, Guangzhou, China Contributions: (I) Conception and design: L Liu, S Xu, Y Weng, S Huang; (II) Administrative support: L Huang, J He; (III) Provision of study materials or patients: L Huang, G Liu; (IV) Collection and assembly of data: S Ma, Y Weng; (V) Data analysis and interpretation: L Liu, S Xu; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Shaohong Huang. Department of Cardiothoracic Surgery, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China. Email: [email protected]; Yimin Weng. Department of Cardiothoracic surgery, Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China. Email: [email protected]. Background: This study applied a complex bioinformatics analysis to explore the hub regulators and immune network to further elucidate the molecular mechanisms of lung adenocarcinoma (LUAD) immune regulation. Methods: LUAD immunological microenvironment features and microenvironment-related differential expression genes (DEGs) were identified by ESTIMATE algorithm and linear models for microarray analyses (LIMMA), respectively. CIBERSORT and Igraph algorithms were applied to construct the LUAD- related immunocyte infiltration and regulatory network. -

TLR8, TLR9 and Gfi-1 Restrain TLR7-Mediated Lupus
Université de la Méditerranée Aix-Marseille II Faculté des Sciences de Luminy Ecole Doctorale des Sciences de la Vie et de la Santé Thèse Pour obtenir le grade de Docteur de l’Université d’Aix-Marseille II Discipline : Immunologie Présenté et soutenue publiquement par Amanda Beatriz Rodrigues Barreto de Macedo TLR8, TLR9 and Gfi-1 restrain TLR7-mediated lupus Directrice de thèse : Lena Alexopoulou Soutenue le 17 novembre 2014, devant le jury composé de : Pr Franck Galland Président Dr Sandra Diebold Rapporteur Dr Claire Lugnier Rapporteur Dr Yannis Morel Examinateur Dr Nathalie Lambert Examinateur Dr Lena Alexopoulou Directrice de thèse Résumé Le lupus érythémateux disseminé (LED) est une maladie chronique auto-immune caractérisée par la production d’autoanticorps dirigés contre les antigènes nucléaires. L’étiologie du lupus n’est pas encore claire mais de nombreuses études indiquent un rôle des récepteurs Toll-like (TLR) et leurs voies de signalisation dans l’initiation et la mise en place du LED. Mon projet de thèse avait deux objectifs dont le premier était de comprendre comment le TLR8 et TLR9 contribuent au lupus dépendant de TLR7. Des études antérieures de notre laboratoire ont révélé que le TLR8 murin contrôle la fonction de TLR7 dans les cellules dendritiques et est aussi impliqué dans le lupus. D’autre part, TLR9 contrôle également le lupus dépendant de TLR7. Cependant, leur contribution respective dans le contrôle de TLR7 restait inconnue. En étudiant des souris doublement déficientes pour TLR8 et TLR9, nous avons constaté que les souris TLR8/9-/- présentent une maladie exacerbée avec une splénomégalie accrue, des niveaux d’autoanticorps circulants plus élevés et des dépôts de complexes immuns plus importants au niveau des reins par rapport aux souris déficientes pour TLR8 ou TLR9 uniquement.