Predicting Mirna Targets for Hepatocellular Carcinoma with an Integrated Method
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Noncoding Rnas As Novel Pancreatic Cancer Targets
NONCODING RNAS AS NOVEL PANCREATIC CANCER TARGETS by Amy Makler A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, FL August 2018 Copyright 2018 by Amy Makler ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous support, constant encouragement, and his gentle, but sometimes critical, guidance throughout the past two years of my master’s education. His faith in my abilities and his belief in my future success ensured I continue down this path of research. Working in Dr. Narayanan’s lab has truly been an unforgettable experience as well as a critical step in my future endeavors. I would also like to extend my gratitude to my committee members, Dr. Binninger and Dr. Jia, for their support and suggestions regarding my thesis. Their recommendations added a fresh perspective that enriched our initial hypothesis. They have been indispensable as members of my committee, and I thank them for their contributions. My parents have been integral to my successes in life and their support throughout my education has been crucial. They taught me to push through difficulties and encouraged me to pursue my interests. Thank you, mom and dad! I would like to thank my boyfriend, Joshua Disatham, for his assistance in ensuring my writing maintained a logical progression and flow as well as his unwavering support. He was my rock when the stress grew unbearable and his encouraging words kept me pushing along. -

Bioinformatics Mining of the Dark Matter Proteome For
BIOINFORMATICS MINING OF THE DARK MATTER PROTEOME FOR CANCER TARGETS DISCOVERY by Ana Paula Delgado A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, Florida May 2015 Copyright 2015 by Ana Paula Delgado ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous encouragement, guidance, and support during the past two years of my graduate education. It has truly been an unforgettable experience working in his laboratory. I also want to express gratitude to my external advisor Professor Van de Ven from the University of Leuven, Belgium for his constant involvement and assistance on my project. Moreover, I would like to thank Dr. Binninger and Dr. Dawson-Scully for their advice and for agreeing to serve on my thesis committee. I also thank provost Dr. Perry for his involvement in my project. I thank Jeanine Narayanan for editorial assistance with the publications and with this dissertation. It has been a pleasure working with various undergraduate students some of whom became lab mates including Pamela Brandao, Maria Julia Chapado and Sheilin Hamid. I thank them for their expert help in the projects we were involved in. Lastly, I want to express my profound thanks to my parents and brother for their unconditional love, support and guidance over the last couple of years. They were my rock when I was in doubt and never let me give up. I would also like to thank my boyfriend Spencer Daniel and best friends for being part of an incredible support system. -

Supplementary Data
Gene expression profile of the A549 human non-small cell lung carcinoma cell line following treatment with the seeds of Descurainia sophia, a potential anticancer drug Bu-Yeo Kim, Jun Lee, Sung Joon Park, Ok-Sun Bang, and No Soo Kim Supplementary Data Figure Legends Supplementary Figure 1. Electropherogram of total RNA samples used for gene expression profiles. The A549 cells were treated with increasing concentrations of EEDS (0-20 mg/mL). After 24 h drug treatment, total RNAs were extracted from the cells, and their integrities were determined by electropherogram. RIN values range from 10 (intact) to 1(totally degraded). Supplementary Figure 2. Pathways enriched (FDR<0.01) in the Up- and Down-patterns. The genes that are shown in Table 5 and Figure 4, and involved in each pathway are highlighted in green Supplementary Figure 3. The level of similarity is represented in red with a scale bar. The color in the box of a diagonal line or “Activity” on the right panel represents the activity of the pathway. The positions and names of signaling-related pathways are colored red and the metabolism-related pathways are colored blue. Supplementary Table 1. Quality control of total RNA samples used for gene expression analysis. * EEDS (mg/mL) OD260/280 OD260/230 Ratio (28s/18s) RIN Result 0 2.04 2.22 2.3 9.8 Pass 1.25 2.03 2.28 2.2 10.0 Pass 5 2.03 2.28 2.1 10.0 Pass 20 2.04 2.23 2.0 10.0 Pass *RIN values, 10 (intact) to 1 (completely degraded). -

Deciphering Common and Rare Genetic E Ects on Reading Ability
Deciphering common and rare genetic eects on reading ability © 2016, Amaia Carrion´ Castillo ISBN: 978-90-76203-78-2 Printed and bound by Ipskamp Drukkers Deciphering common and rare genetic eects on reading ability Proefschri ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnicus prof. dr. J.H.J.M. van Krieken, volgens besluit van het college van decanen in het openbaar te verdedigen op maandag 7 november 2016 om 12.30 uur precies door Amaia Carrion´ Castillo geboren op 22 november 1988 te Donostia (Spanje) Promotor Prof. dr. Simon E. Fisher Copromotor Dr. Clyde Francks (MPI) Manuscriptcommissie Prof. dr. Anneke I. den Hollander Prof. dr. Juha Kere (Karolinska Institutet, Zweden) Prof. dr. Gerd Schulte-Korne¨ (Ludwig-Maximilians-University Munich, Duitsland) Deciphering common and rare genetic eects on reading ability Doctoral esis to obtain the degree of doctor from Radboud University Nijmegen on the authority of the Rector Magnicus prof. dr. J.H.J.M. van Krieken, according to the decision of the Council of Deans to be defended in public on Monday, November 7, 2016 at 12.30 hours by Amaia Carrion´ Castillo Born on 22 November 1988 in Donostia (Spain) Supervisor Prof. dr. Simon E. Fisher Co-supervisor Dr. Clyde Francks (MPI) Doctoral esis Committee Prof. dr. Anneke I. den Hollander Prof. dr. Juha Kere (Karolinska Institutet, Sweden) Prof. dr. Gerd Schulte-Korne¨ (Ludwig-Maximilians-University Munich, Germany) Etxekoei CONTENTS 1 General introduction 11 1.1 e learning-to-read brain ........................ 11 1.2 Variation in reading ability........................ 13 1.3 Complex and multifactorial aetiology................. -
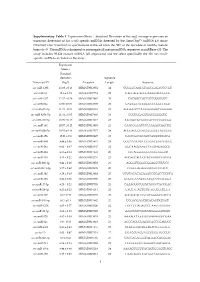
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Position Effect on FGF13 Associated with X-Linked Congenital Generalized Hypertrichosis
Position effect on FGF13 associated with X-linked congenital generalized hypertrichosis Gina M. DeStefanoa, Katherine A. Fantauzzoa,b, Lynn Petukhovab,c, Mazen Kurbanb, Marija Tadin-Strappsa, Brynn Levyd, Dorothy Warburtona,e, Elizabeth T. Cirullif, Yujun Hanf, Xiaoyun Sung, Yufeng Sheng, Maryam Shirazid, Vaidehi Jobanputrad, Rodrigo Cepeda-Valdesh, Julio Cesar Salas-Alanish,i, and Angela M. Christianoa,b,1 Departments of aGenetics and Development, bDermatology, cEpidemiology, and dPathology and Cell Biology, Columbia University, New York, NY; eDepartment of Pediatrics, Columbia University Medical Center, New York, NY 10032; fCenter for Human Genome Variation, Duke University School of Medicine, Durham, NC 27708; gDepartment of Biomedical Informatics, Columbia Initiative in Systems Biology, Columbia University, New York, NY 10032; hDystrophic Epidermolysis Bullosa Research Association (DebRA), Nuevo Leon 67150, Mexico; and iBasic Science, Universidad de Monterrey, Nueva Leon 63238, Mexico Edited by Elaine Fuchs, The Rockefeller University, New York, NY, and approved March 12, 2013 (received for review October 17, 2012) X-linked congenital generalized hypertrichosis (Online Mendelian providing evidence to support a position effect as the un- Inheritance in Man 307150) is an extremely rare condition of hair derlying genetic basis of X-linked hypertrichosis. overgrowth on different body sites. We previously reported link- age in a large Mexican family with X-linked congenital generalized Results and Discussion hypertrichosis cosegregating with deafness and with dental and We ascertained a large kindred from Mexico with X-linked CGH (Online Mendelian Inheritance in Man 307150) cosegregating palate anomalies to Xq24-27. Using SNP oligonucleotide microar- – ray analysis and whole-genome sequencing, we identified a 389- with deafness and with dental and palate anomalies (Fig. -
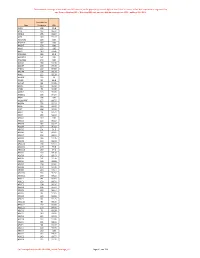
Onderstaande Coverage Is Berekend Over 860 Exomen, Welke Geprept Zijn Met De Agilent Sureselect XT Exome V7 Kit
Onderstaande coverage is berekend over 860 exomen, welke geprept zijn met de Agilent SureSelect XT exome v7 kit. Het sequencen is uitgevoerd op een Illumina NextSeq500 of NovaSeq6000 met een gemiddelde coverage van 100X , dekking 20x >93%. Gemiddelde Gen Coverage 20x A1BG 128 99.8 A1CF 132 99.32 A2ML1 155 99.97 A2M 148 99.72 A3GALT2 119 100 A4GALT 157 100 A4GNT 178 100 AAAS 123 100 AACS 143 97.96 AADACL2 186 99.9 AADACL3 161 100 AADACL4 170 100 AADAC 160 99.98 AADAT 138 97.39 AAED1 110 84.89 AAGAB 164 99.29 AAK1 122 99.39 AAMDC 135 90 AAMP 95 98.4 AANAT 86 99.85 AAR2 120 98.41 AARD 75 99.06 AARS2 123 99.89 AARSD1 115 94.24 AARS 134 100 AASDHPPT 157 99.75 AASDH 135 99.29 AASS 143 99.67 AATF 156 99.95 AATK 79 92.71 ABAT 136 99.16 ABCA1 149 100 ABCA2 123 96.52 ABCA3 132 99.27 ABCA4 137 96.03 ABCA5 101 94.3 ABCA6 131 98.41 ABCA7 102 99.12 ABCA8 146 99.26 ABCA9 140 99.34 ABCA10 126 94.11 ABCA12 159 99.8 ABCA13 159 97.2 ABCB1 160 99.58 ABCB4 142 99.43 ABCB5 151 99.76 ABCB6 149 99.98 ABCB7 128 99.81 ABCB8 100 93.36 ABCB9 124 97.14 ABCB10 136 90.97 ABCB11 158 99.22 ABCC1 141 96.65 ABCC2 150 99.71 ABCC3 108 96.16 ABCC4 134 95.97 ABCC5 130 96.14 ABCC6 96 92.85 ABCC8 136 99.83 ABCC9 141 99.75 ABCC10 90 99.35 ABCC11 117 99.93 ABCC12 153 99.93 ABCD1 74 83.58 ABCD2 143 99.92 ABCD3 122 92.63 ABCD4 122 99.84 ABCE1 141 98.18 ABCF1 90 99.42 ABCF2 128 99.93 ABCF3 159 100 ABCG1 135 99.71 ABCG2 143 99.77 ABCG4 122 99.91 De Coverage komt vanuit LAB-F0680_Exoom Coverage_v3. -

Genome-Scale Detection of Positive Selection in 9 Primates Predicts Human-Virus Evolutionary Conflicts
bioRxiv preprint doi: https://doi.org/10.1101/131680; this version posted April 27, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Genome-scale detection of positive selection in 9 primates predicts human-virus evolutionary conflicts Robin van der Lee1,*,^, Laurens Wiel1,2, Teunis J.P. van Dam1,#, Martijn A. Huynen1 1Centre for Molecular and Biomolecular Informatics, 2Department of Human Genetics, Radboud Institute for Molecular Life Sciences, Radboud university medical center, Nijmegen, The Netherlands *Correspondence: [email protected] ^Current address: Centre for Molecular Medicine and Therapeutics, Department of Medical Genetics, BC Children’s Hospital Research Institute, University of British Columbia, Vancouver, Canada #Current address: Theoretical Biology and Bioinformatics, Department of Biology, Faculty of Science, Utrecht University, The Netherlands bioRxiv preprint doi: https://doi.org/10.1101/131680; this version posted April 27, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Hotspots of rapid genome evolution hold clues about human adaptation. Here, we present a comparative analysis of nine whole-genome sequenced primates to identify high-confidence targets of positive selection. We find strong statistical evidence for positive selection acting on 331 protein-coding genes (3%), pinpointing 934 adaptively evolving codons (0.014%). -

Mass Spectrometry-Based Proteomics Analysis of Bioactive Proteins in EMD That Modulate Adhesion of Gingival Fibroblast to Improve Bio-Integration of Dental Implants
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 3-28-2019 3:00 PM Mass Spectrometry-Based Proteomics Analysis Of Bioactive Proteins In EMD That Modulate Adhesion Of Gingival Fibroblast To Improve Bio-Integration Of Dental Implants David Zuanazzi Machado Jr The University of Western Ontario Supervisor Siqueira, Walter L. The University of Western Ontario Graduate Program in Biochemistry A thesis submitted in partial fulfillment of the equirr ements for the degree in Doctor of Philosophy © David Zuanazzi Machado Jr 2019 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Biochemistry Commons, Dental Materials Commons, Oral Biology and Oral Pathology Commons, and the Periodontics and Periodontology Commons Recommended Citation Zuanazzi Machado, David Jr, "Mass Spectrometry-Based Proteomics Analysis Of Bioactive Proteins In EMD That Modulate Adhesion Of Gingival Fibroblast To Improve Bio-Integration Of Dental Implants" (2019). Electronic Thesis and Dissertation Repository. 6064. https://ir.lib.uwo.ca/etd/6064 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. i ABSTRACT Titanium (Ti) implants are used in dental practice to replace damaged or lost teeth. For effective treatment, the dental implant needs to integrate with the surrounding hard and soft tissues on the implant site. Despite improvements in bone-implant integration that have been achieved through surface modifications, the integration with the soft tissues is still deficient. The oral mucosa that embraces the transmucosal component of the implant only contacts the surface without making a strong attachment with the connective tissue. -

Syndromic Cases of Hemophilia B and Morbid Obesity Due to Contiguous
Syndromic cases of hemophilia B and morbid obesity due to contiguous gene deletions on Xq26.3-q27.2: unsuspected phenotype-genotype associations by bioinformatics and extensive clinical data mining Pamela Radic1, Miguel Abelleyro1, Betiana Ziegler1, Vanina Marchione1, Julian Nevado2, Pablo Lapunzina2, Gabriela Sciuccati3, Daniela Neme4, Liliana Rossetti1, Mariana Bonduel5, and Carlos De Brasi1 1Instituto de Medicina Experimental (IMEX-CONICET/ANM) 2Instituto de Gen´eticaM´edicay Molecular (INGEMM)-IdiPAZ, Hospital Universitario La Paz 3Hospital de Pediatr´ıaProf Dr Juan P Garrahan Servicio de Hematolog´ıay Oncolog´ıa 4Fundaci´onde la Hemofilia Alfredo Pavlovsky. 5Hospital de Pediatria Prof Dr Juan P Garrahan Servicio de Hematologia y Oncologia August 20, 2020 Abstract Hemophilia B (HB) associates with pathogenic F9-variants. The literature showed that hemizygous deletions encompassing F9 and vicinal genes may express extra-phenotypes suggesting new causal relationships. Aim: Analyze the molecular basis of syndromic cases of HB, obesity (OB), severe global developmental delay (SGDD) and generalized hypotonia (GH). Whole F9-deletions were detected in 3 hemizygous probands with HB. Dense SNP-array and case-specific STS walking strategies al- lowed amplification and characterization of the deletion breakpoints. Bioinformatic/statistical analyses included data mining in HPO (Human Phenotype Ontology), OMIM, STRING (protein-protein interaction networks) databases and estimation of null-hypothesis-based Expected-values. Patients (cases#3-case#1) showed complete F9 deletions involving 0.16-4.34-Mb and 1- 17 additional genes on Xq26.3-q27.2. Bioinformatic/statistical approaches revealed highly significant STRING-associations (P[?]0.00115) between case#1/#2 common deleted genes (SOX3, FGF13, CXorf66) and those HPO associated with OB (20/343), GH (36/923) and SGDD (10/119). -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -

A Novel Transmembrane Glycoprotein Cancer Biomarker Present in the X Chromosome ANA PAULA DELGADO, SHEILIN HAMID, PAMELA BRANDAO and RAMASWAMY NARAYANAN
CANCER GENOMICS & PROTEOMICS 11 : 81-92 (2014) A Novel Transmembrane Glycoprotein Cancer Biomarker Present in the X Chromosome ANA PAULA DELGADO, SHEILIN HAMID, PAMELA BRANDAO and RAMASWAMY NARAYANAN Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, Boca Raton, FL, U.S.A. Abstract. Background: The uncharacterized proteins of the genome are of unknown nature (10). These proteins and the human proteome offer an untapped potential for cancer non-coding RNAs are called the ‘dark matter’ of the genome biomarker discovery. Numerous predicted open reading (11-13). Whereas in the past most gene discovery has frames (ORFs) are present in diverse chromosomes. The revolved around known genes due to the ease of follow-up mRNA and protein expression data, as well as the mutational studies (14-17), the dark matter of the genome offers an and variant information for these ORF proteins are available untapped potential (18). Realizing the importance of this area, in the cancer-related bioinformatics databases. Materials the US National Cancer Institute has recently announced a and Methods: ORF proteins were mined using bioinformatics major initiative called illuminating the dark matter for and proteomic tools to predict motifs and domains, and druggable targets (http://commonfund.nih.gov/idg/index). cancer relevance was established using cancer genome, Establishing cancer relevance for novel or uncharacterized transcriptome and proteome analysis tools. Results: A novel proteins is a crucial first step in lead discovery. The cancer testis-restricted ORF protein present in chromosome X called genomes of patients from around the world can be readily CXorf66 was detected in the serum, plasma and neutrophils.