Case Report AJNT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hypokalaemia in a Woman with Eating Disorder
Grand Rounds Vol 11 pages 53–55 Specialities: Acute Medicine; Nephrology; Psychiatry Article Type: Case Report DOI: 10.1102/1470-5206.2011.0013 ß 2011 e-MED Ltd Hypokalaemia in a woman with eating disorder Zachary Z. Brenera, Boris Medvedovskya, James F. Winchestera and Michael Bergmanb aDivision of Nephrology, Department of Medicine, Beth Israel Medical Center, Albert Einstein School of Medicine of Yeshiva University, New York, USA; bDepartment of Medicine, Campus Golda, Rabin Medical Center, Petah-Tikva, Tel-Aviv University, Israel Corresponding address: Dr Zachary Z. Brener, 350 E. 17th St., Division of Nephrology, Beth Israel Medical Center, New York, NY 10003, USA. Email: [email protected] Date accepted for publication 13 April 2011 Abstract Chronic hypokalaemia often remains a diagnostic challenge, especially in young women without hypertension. A concealed diuretic abuse should be suspected, especially in young women with eating disorders. This case describes a woman with chronic hypokalaemia in whom a thorough medical history and proper laboratory tests were essential to early and accurate diagnosis. Keywords Hypokalaemia; eating disorders; diuretics. Introduction Chronic hypokalaemia often remains a diagnostic challenge, especially in young women without hypertension. After the exclusion of the most obvious causes, a concealed diuretic abuse associated with or without surreptitious vomiting and laxative abuse should be suspected, especially in young women concerned with their body image. A conclusive diagnosis may be difficult as such patients often vigorously deny diuretic intake[1]. Also, only a minority of patients with eating disorders (approximately 6%) abuse diuretics[2–4]. This case describes a woman with chronic hypokalaemia in whom a thorough medical history and proper laboratory tests were essential to an early and accurate diagnosis. -

Magnesium: the Forgotten Electrolyte—A Review on Hypomagnesemia
medical sciences Review Magnesium: The Forgotten Electrolyte—A Review on Hypomagnesemia Faheemuddin Ahmed 1,* and Abdul Mohammed 2 1 OSF Saint Anthony Medical Center, 5666 E State St, Rockford, IL 61108, USA 2 Advocate Illinois Masonic Medical Center, 833 W Wellington Ave, Chicago, IL 60657, USA; [email protected] * Correspondence: [email protected] Received: 20 February 2019; Accepted: 2 April 2019; Published: 4 April 2019 Abstract: Magnesium is the fourth most abundant cation in the body and the second most abundant intracellular cation. It plays an important role in different organ systems at the cellular and enzymatic levels. Despite its importance, it still has not received the needed attention either in the medical literature or in clinical practice in comparison to other electrolytes like sodium, potassium, and calcium. Hypomagnesemia can lead to many clinical manifestations with some being life-threatening. The reported incidence is less likely than expected in the general population. We present a comprehensive review of different aspects of magnesium physiology and hypomagnesemia which can help clinicians in understanding, identifying, and treating this disorder. Keywords: magnesium; proton pump inhibitors; diuretics; hypomagnesemia 1. Introduction Magnesium is one of the most abundant cation in the body as well as an abundant intracellular cation. It plays an important role in molecular, biochemical, physiological, and pharmacological functions in the body. The importance of magnesium is well known, but still it is the forgotten electrolyte. The reason for it not getting the needed attention is because of rare symptomatology until levels are really low and also because of a lack of proper understanding of magnesium physiology. -
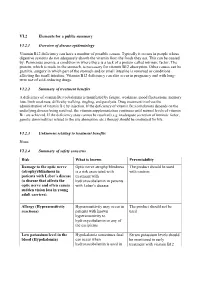
VI.2 Elements for a Public Summary VI.2.1 Overview of Disease Epidemiology Vitamin B12 Deficiency Can Have a Number of Possible
VI.2 Elements for a public summary VI.2.1 Overview of disease epidemiology Vitamin B12 deficiency can have a number of possible causes. Typically it occurs in people whose digestive systems do not adequately absorb the vitamin from the foods they eat. This can be caused by: Pernicious anemia, a condition in where there is a lack of a protein called intrinsic factor. The protein, which is made in the stomach, is necessary for vitamin B12 absorption. Other causes can be gastritis, surgery in which part of the stomach and/or small intestine is removed or conditions affecting the small intestine. Vitamin B12 deficiency can also occur in pregnancy and with long- term use of acid-reducing drugs. VI.2.2 Summary of treatment benefits A deficiency of vitamin B12 (cobalamin) is manifested by fatigue, weakness, mood fluctuations, memory loss, limb weakness, difficulty walking, tingling, and paralysis. Drug treatment involves the administration of vitamin B12 by injection. If the deficiency of vitamin B12 (cobalamin) depends on the underlying disease being resolved, the vitamin supplementation continues until normal levels of vitamin B12 are achieved. If the deficiency state cannot be resolved (e.g. inadequate secretion of intrinsic factor, genetic abnormalities related to the site absorption, etc.) therapy should be continued for life. VI.2.3 Unknowns relating to treatment benefits None. VI.2.4 Summary of safety concerns Risk What is known Preventability Damage to the optic nerve Optic nerve atrophy/blindness The product should be used (atrophy)/blindness in is a risk associated with with caution. patients with Leber´s disease treatment with (a disease that affects the hydroxocobalamin in patients optic nerve and often causes with Leber´s disease. -

Mechanism of Hypokalemia in Magnesium Deficiency
SCIENCE IN RENAL MEDICINE www.jasn.org Mechanism of Hypokalemia in Magnesium Deficiency Chou-Long Huang*† and Elizabeth Kuo* *Department of Medicine, †Charles and Jane Pak Center for Mineral Metabolism and Clinical Research, University of Texas Southwestern Medical Center, Dallas, Texas ABSTRACT deficiency is likely associated with en- ϩ Magnesium deficiency is frequently associated with hypokalemia. Concomitant hanced renal K excretion. To support magnesium deficiency aggravates hypokalemia and renders it refractory to treat- this idea, Baehler et al.5 showed that ad- ment by potassium. Herein is reviewed literature suggesting that magnesium ministration of magnesium decreases ϩ deficiency exacerbates potassium wasting by increasing distal potassium secre- urinary K excretion and increases se- ϩ tion. A decrease in intracellular magnesium, caused by magnesium deficiency, rum K levels in a patient with Bartter releases the magnesium-mediated inhibition of ROMK channels and increases disease with combined hypomagnesemia potassium secretion. Magnesium deficiency alone, however, does not necessarily and hypokalemia. Similarly, magnesium ϩ cause hypokalemia. An increase in distal sodium delivery or elevated aldosterone replacement alone (without K ) in- ϩ levels may be required for exacerbating potassium wasting in magnesium creases serum K levels in individuals deficiency. who have hypokalemia and hypomag- nesemia and receive thiazide treatment.6 J Am Soc Nephrol 18: 2649–2652, 2007. doi: 10.1681/ASN.2007070792 Magnesium administration decreased urinary Kϩ excretion in these individuals (Dr. Charles Pak, personal communica- Hypokalemia is among the most fre- cin B, cisplatin, etc. Concomitant magne- tion, UT Southwestern Medical Center quently encountered fluid and electro- sium deficiency has long been appreci- at Dallas, July 13, 2007). -

An Infant with Chronic Hypernatremia
European Journal of Endocrinology (2006) 155 S141–S144 ISSN 0804-4643 An infant with chronic hypernatremia M L Marcovecchio Department of Paediatrics, University of Chieti, Via dei Vestini 5, 66100 Chieti, Italy (Correspondence should be addressed to M L Marcovecchio; Email: [email protected]) Abstract A 4-month-old boy was presented with failure to thrive, refusal to feed, delayed motor development, truncal hypotonia, and head lag. His plasma osmolality and sodium were significantly high, while his urine osmolality was inappropriately low and did not increase after desmopressin administration. Despite his hyperosmolality, he presented with a lack of thirst and became clearly polyuric and polydipsic only at the age of 2 years. Initial treatment with indomethacin was ineffective, while the combination of hydrochlorothiazide and amiloride was effective and well tolerated. European Journal of Endocrinology 155 S141–S144 Introduction (61 ml/kg) respectively. Other investigations including thyroid and adrenal function were normal. He was Chronic hypernatremia in young patients is generally refusing oral fluids despite being hypernatremic. the result of alterations in the mechanisms controlling A cranial magnetic resonance imaging scan excluded fluid balance (1). Excessive water loss, as in diabetes structural abnormalities in the hypophysis, hypo- insipidus, or an inadequate fluid intake, as in adipsic thalamus and surrounding area. There was no response hypernatremia, may be the underlying cause. The to 0.3 mg 1-desamino-8-D-arginine-vasopressin (DDAVP) differential diagnosis between these conditions is given intravenously (osmolality pre- 374 mosmol/kg; important in order to choose the appropriate treatment. post- 371 mosmol/kg). This suggested a tubular defect However, pitfalls in the diagnosis are often related to an causing nephrogenic diabetes insipidus (NDI). -

Electrolyte and Acid-Base Disorders Triggered by Aminoglycoside Or Colistin Therapy: a Systematic Review
antibiotics Review Electrolyte and Acid-Base Disorders Triggered by Aminoglycoside or Colistin Therapy: A Systematic Review Martin Scoglio 1,* , Gabriel Bronz 1, Pietro O. Rinoldi 1,2, Pietro B. Faré 3,Céline Betti 1,2, Mario G. Bianchetti 1, Giacomo D. Simonetti 1,2, Viola Gennaro 1, Samuele Renzi 4, Sebastiano A. G. Lava 5 and Gregorio P. Milani 2,6,7 1 Faculty of Biomedicine, Università della Svizzera Italiana, 6900 Lugano, Switzerland; [email protected] (G.B.); [email protected] (P.O.R.); [email protected] (C.B.); [email protected] (M.G.B.); [email protected] (G.D.S.); [email protected] (V.G.) 2 Department of Pediatrics, Pediatric Institute of Southern Switzerland, Ospedale San Giovanni, Ente Ospedaliero Cantonale, 6500 Bellinzona, Switzerland; [email protected] 3 Department of Internal Medicine, Ospedale La Carità, Ente Ospedaliero Cantonale, 6600 Locarno, Switzerland; [email protected] 4 Division of Hematology and Oncology, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; [email protected] 5 Pediatric Cardiology Unit, Department of Pediatrics, Centre Hospitalier Universitaire Vaudois, and University of Lausanne, 1011 Lausanne, Switzerland; [email protected] 6 Pediatric Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy 7 Department of Clinical Sciences and Community Health, Università degli Studi di Milano, 20122 Milan, Italy * Correspondence: [email protected] Citation: Scoglio, M.; Bronz, G.; Abstract: Aminoglycoside or colistin therapy may alter the renal tubular function without decreasing Rinoldi, P.O.; Faré, P.B.; Betti, C.; the glomerular filtration rate. This association has never been extensively investigated. -

The Serum Sodium, Potassium and Calcium Levels in Children 6- 59 Months of Age with Severe Acute Malnutrition
ORIGINAL ARTICLE The Serum Sodium, Potassium and Calcium Levels in Children 6- 59 Months of Age with Severe Acute Malnutrition BILQUEES FATIMA1, MUHAMMAD AMIN SHEIKH2, MALIK MUHAMMAD NAEEM3 ABSTRACT Aim: To study the serum sodium, potassium and calcium levels in cases of SAM and their comparison in cases with and without diarrhea Methods: This cross sectional study was conducted in the pediatric unit II, Bahawal Victoria Hospital, Bahawalpur from January 2016 to June 2016 in children having age between 6 to 59 months admitting with a diagnosis of SAM. The patients were divided into two groups, Group A with diarrhea and group B without diarrhea. Blood was taken for serum sodium, potassium and calcium. Results: 100 patients were included. Their mean±SD age in months was 23.56±3.80. 62% were males. Serum sodium (mean±SD) was 138.46±4.14 mmol/L, potassium 3.961±0.691 mmol/L and calcium 8.359±0.61 mg/dl. 67 cases presented with (Group A) while 33 without diarrhea (Group B). The serum sodium (mean±SD) was 138.51±4.22 mmol/L in group A while 138.36± 4.05 in group B (p 0.1), the potassium 3.89±0.67 in group A while 4.1±0.72 in group B (p 0.009) and calcium (mg/dL) 8.39±0.6 in group A while 8.34 ±0.73 mg/dL in group B (0.71). There was isolated hyponatremia in 10.4% cases in group A and 9.1% in group B (p 0.83) while isolated hypokalemia in 26.9% cases in group A and 18.2% in group B (p 0.34). -

Pseudo Bartter Syndrome from Surreptitious Purging Behaviour In
al Dis ion ord rit e t rs u N & f T o h Gentile, J Nutr Disorders Ther 2012, 2:1 l e a r n a r DOI: 10.4172/2161-0509.1000107 p u y o Journal of Nutritional Disorders & Therapy J ISSN: 2161-0509 Case Report Open Access Pseudo Bartter Syndrome from Surreptitious Purging Behaviour in Anorexia Nervosa Maria Gabriella Gentile* Eating Disorder Unit, Niguarda Hospital, Piazza Ospedale Maggiore 3, 20126 Milan, Italy Abstract Pseudo Bartter syndrome is a rare disorder characterized by metabolic alkalosis, hypokalaemia, hyperaldosteronism, hyperreninism, normal blood pressure and hyperplasia of the juxtaglomerular apparatus. The most dangerous complication of Pseudo Bartter syndrome is hypokalemia. Hypokalemia caused by vomiting, diarrhea, prolonged fasting, abuse of potassium-depleting drugs, may be present in patients with binge /purging form of anorexia or bulimia nervosa. We report a case of a 19-year-old girl with anorexia nervosa (BMI 16.15 kg/m2) and severe prolonged hypokalemia (1.9 mEq/l), metabolic alkalosis and severe protracted secondary hyperaldosteronism (i.e. Pseudo Bartter’s syndrome) from surreptitious purging behaviour (vomit and laxative abuse). An intensive multidisciplinary day-hospital treatment including long-term potassium supplementation, a potassium- sparing diuretic was necessary to resolve the case and to allow the young girl to admit her previous purging behaviour and after three months to get at a normal kalemia without any potassium supplementation and BMI at a normal value (20 kg/m2). Given the dangers to the heart electrical and mechanical functions set by severe potassium deficiency, it is mandatory to find out the true cause so that a proper treatment can be started. -

Qtc Prolongation Is Associated with Hypokalemia and Hypocalcemia in Emergency Department Patients Lucy Franjic Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Division of Emergency Medicine/Emergency Care Conference Abstracts and Posters Research Section 2012 QTc prolongation is associated with hypokalemia and hypocalcemia in emergency department patients Lucy Franjic Washington University School of Medicine in St. Louis Stacey House Washington University School of Medicine in St. Louis Irena Vitkovitsky Washington University School of Medicine in St. Louis S. Eliza Halcomb Washington University School of Medicine in St. Louis Follow this and additional works at: http://digitalcommons.wustl.edu/em_conf Recommended Citation Franjic, Lucy; House, Stacey L.; Vitkovitsky, Irena; Halcomb, S. Eliza, "QTc prolongation is associated with hypokalemia and hypocalcemia in emergency department patients" (2012). Conference Abstracts and Posters. Paper 12. http://digitalcommons.wustl.edu/em_conf/12 This Presentation Paper is brought to you for free and open access by the Division of Emergency Medicine/Emergency Care Research Section at Digital Commons@Becker. It has been accepted for inclusion in Conference Abstracts and Posters by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. QTc Prolongation is Associated with Hypokalemia and Hypocalcemia in Emergency Department Patients Lucy Franjic, MD Stacey L. House MD PhD, Lucy Franjic MD, Irena Vitkovitsky MD, S. Eliza Halcomb MD Washington University in St. Louis Division of Emergency Medicine Society for Academic Emergency Medicine Great Plains Regional Research Forum St. Louis, MO. September 2012 © Stacey House, 2012 QTc Prolongation Congenital Six types (LQT1-LQT6) Mutations in genes encoding potassium and sodium transmembrane channel proteins Acquired Hypokalemia, hypocalcemia, hypomagnesemia, HIV, myocardial ischemia, numerous medications and drugs (i.e. -
Renal Tubular Acidosis Presenting with Progressive Gross Motor Developmental Regression and Acute Paralysis Randula Ranawaka1*, Kavinda Dayasiri2 and Manoji Gamage3
Ranawaka et al. BMC Res Notes (2017) 10:618 DOI 10.1186/s13104-017-2949-2 BMC Research Notes CASE REPORT Open Access A child with distal (type 1) renal tubular acidosis presenting with progressive gross motor developmental regression and acute paralysis Randula Ranawaka1*, Kavinda Dayasiri2 and Manoji Gamage3 Abstract Background: Distal (Type 1) renal tubular acidosis (dRTA) is characterized by inability to secrete hydrogen irons from the distal tubule. The aetiology of dRTA is diverse and can be either inherited or acquired. Common clinical presenta- tions of dRTA in the paediatric age group include polyuria, nocturia, failure to thrive, constipation, abnormal breath- ing and nephrolithiasis. Though persistent hypokalemia is frequently seen in dRTA, hypokalemic muscular paralysis is uncommon and rarely described in children. Case presentation: Three and a half years old girl was referred for evaluation of progressive loss of gross motor milestones over 6 months and acute episode of paralysis. Her other developmental domains were age appropriate. Notably, there was no history of polyuria, polydipsia, nocturia and abnormal breathing. Physical examination revealed proximal myopathy (waddling gait and positive Gower’s sign), diminished lower limb refexes and muscle tone. Her serum potassium was low (2.1 meq/l) and she was subsequently investigated for hypokalemic paralysis. Diagnosis of distal renal tubular acidosis was made, based on hypokalemic hyperchloremic metabolic acidosis with normal anion gap, high urine pH, borderline hypercalciuria, medullary nephrocalcinosis and exclusion of other diferential diagno- sis. The child showed complete symptomatic recovery upon commencement of standard treatment for distal renal tubular acidosis. Conclusions: This case report highlights the importance of considering hypokalemia and renal tubular acidosis in the diferential diagnosis of acute faccid paralysis and proximal myopathy. -

A Systematic Review of the Role of Thiamine Supplementation in Treatment of Refeeding Syndrome
Top Clin Nutr Vol. 36, No. 1, pp. 36–51 Copyright c 2021 Wolters Kluwer Health, Inc. All rights reserved. NARRATIVE REVIEW A Systematic Review of the Role of Thiamine Supplementation in Treatment of Refeeding Syndrome Lea Steiner, MS, RD, CSG; Susan Hewlings, PhD, RD The purpose of this systematic review is to identify studies where measurable thiamine supple- mentation was provided to patients at risk for or with refeeding syndrome to improve treatment guidelines. A systematic review of PubMed and CINAHL Plus databases was conducted using the terms refeeding syndrome, hypophosphatemia, thiamine, and vitamin B1. A total of 173 articles were retrieved and 11 case studies and 1 retrospective study met inclusion criteria. All studies identified symptoms of thiamine deficiency, and all studies indicated thiamine supplementation was associated with improved clinical symptoms and no harmful outcomes. Average dose provided was 173-mg thiamine/day. Key words: B1, hypophosphatemia, refeeding, refeeding syndrome, thiamine EFEEDING syndrome is defined as the case studies in eating disorders or prolonged R metabolic process in response to the voluntary fasts6 where refeeding syndrome is reintroduction of calories after starvation.1-3 self-induced because ethical barriers inhibit It is often diagnosed by electrolyte abnormal- controlled trials. Related to the paucity of ities and poor management can be fatal.1,2,4 studies and lack of comparability within the Refeeding syndrome was first recognized research available, evidence-based treatment in prisoners -

Evaluating and Managing Electrolyte Disbalances in the Outpatient Setting Disclosure
EVALUATING AND MANAGING ELECTROLYTE DISBALANCES IN THE OUTPATIENT SETTING DISCLOSURE There are no conflicts of interest Homeostasis • Potassium • Water Evaluation and Management of Electrolyte TO BE DISCUSSED Disbalances • Hyperkalemia • Hypokalemia • Hypernatremia • Hyponatremia Summary DISORDERS OF POTASSIUM BALANCE: HYPERKALEMIA AND HYPOKALEMIA POTASSIUM HOMEOSTASIS • Aldosterone • High Na+ delivery to distal tubule Increase (diuretics) Renal K+ • High urine flow (osmotic diuresis) Excretion • High serum K+ level • Delivery bicarbonate to distal tubule • Absence, or very low aldosterone • Low Na+ delivery to the distal Decrease tubule Renal K+ • Low urine flow Excretion • Low serum K+ level • Kidney Injury MORTALITY IN DYSKALEMIA Collins et al Am J Nephrol 2017;46:213- 221 HYPERKALEMIA MY PATIENT HAS HYPERKALEMIA, WHAT SHOULD I DO? H&P and Check other medication causes •Changes? Treat review •Send home •Symptoms? Treat •Metabolic acidosis •Obstruction •Hyperosmolarity •K+ increasing meds •CKD Pseudohyperkale EKG and BMP mia CAUSES OF HYPERKALEMIA Increased potassium release from Reduced urinary potassium excretion cells • Pseudohyperkalemia • Acute and chronic kidney disease • Fist clenching • Reduced aldosterone secretion or • Tourniquet use response to aldosterone • Metabolic acidosis • Reduced distal sodium and water • Insulin deficiency, hyperglycemia, delivery and hyperosmolality • Drugs • Increased tissue catabolism • Drugs • Hyperkalemic Periodic Palasysis Biff Palmer A Physiologic-Based Approach to the Evaluation of a Patient