Purification of Recombinant Proteins by Chemical Removal of the Affinity Tag
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Advances in Cyanamide Chemistry: Synthesis and Applications
molecules Review Recent Advances in Cyanamide Chemistry: Synthesis and Applications M. R. Ranga Prabhath, Luke Williams, Shreesha V. Bhat and Pallavi Sharma * School of Chemistry, Joseph Banks Laboratories, University of Lincoln, Lincoln LN6 7DL, UK; [email protected] (M.R.R.P.); [email protected] (L.W.); [email protected] (S.V.B.) * Correspondence: [email protected]; Tel.: +44-015-2288-6885 Academic Editor: Margaret A. Brimble Received: 9 March 2017; Accepted: 7 April 2017; Published: 12 April 2017 Abstract: The application of alkyl and aryl substituted cyanamides in synthetic chemistry has diversified multi-fold in recent years. In this review, we discuss recent advances (since 2012) in the chemistry of cyanamides and detail their application in cycloaddition chemistry, aminocyanation reactions, as well as electrophilic cyanide-transfer agents and their unique radical and coordination chemistry. Keywords: cyanamide; synthesis; aminocyanation; cycloaddition; electrophilic cyanation; radical reaction; coordination chemistry 1. Introduction Cyanamide enjoys a rich chemical history, which can be traced to its unique and chemically promiscuous nitrogen-carbon-nitrogen (NCN) connectivity. The chemistry of the nitrile-substituted amino-group of the ‘cyanamide-moiety’ is dominated by an unusual duality of a nucleophilic sp3-amino nitrogen and an electrophilic nitrile unit. The reported use of unsubstituted cyanamide (NH2CN) and metal cyanamides (MNCN, where M = metal) date back as far as the late 19th century, where the likes of calcium cyanamide (CaNCN) was used as a fertilizer, and later as source of ammonia and nitric acid, which fueled the industrial production of metal cyanamides. In contrast, the reported use of the corresponding substituted organic cyanamides (RNHCN or RR’NCN) gathered pace only in more recent years. -

Cyanogera Bromide and Cyanogen. by AUGUSTUSEDWARD DIXON and JOHNTAYLOR
View Article Online / Journal Homepage / Table of Contents for this issue 974 DIXON AND TAYLOR: GI.- Cyanogera Bromide and Cyanogen. By AUGUSTUSEDWARD DIXON and JOHNTAYLOR. CYANOGENbromide, in cold aqueous solution, or in the presence of such dilute acids as do not of themselves chemically decompose it, shows no evidence of suffering ionic dissociation. The dilute aqueous solution has the same odour as the solid compound; even after long keeping it yields with silver nitrate no turbidity; it is neutral to litmus, and the pungent vapour fails to give the guaiacum and copper sulphate reaction for hydrogen cyanide ; moreover, the solution is a very feeble conductor of electricity. Although the mixture produced by treating cyanogen bromide with alkali hydroxide contains but alkali bromide and alkali cyanate, Chattaway and Wadmore are of opinion (T., 1902, 81, 199) that hypobromite must first be formed, and then reduced. That cyanate is not directly formed in the reaction with alkali hydroxide is proved from the following facts: (1) Alkali cyanate is not reduced to cyanide by hydriodic acid, ferrous sulphate and alkali, sulphurous acid, alkali sulphite, or even by treatment with aluminium and alkali hydroxide. Further, it has no action on carbamide, either alone or in presence of alkali, (2) If cyanogen bromide is treated with alkali iodide, followed -by alkali, the mixture contains cyanide, but no cyanate, and, when acidified, yields free iodine. (3) If it is treated with ferrous sulphate, and subsequently with alkali and ferric salt, the mixture on acidification gives Prussian- blue, but contains no cyanate. Published on 01 January 1913. -

Reverse-Phase High-Performance Liquid Chromatography of Hydrophobic Proteins and Fragments Thereof’
ANALYTICAL BIOCHEMISTRY 131,99-107 (1983) Reverse-Phase High-Performance Liquid Chromatography of Hydrophobic Proteins and Fragments Thereof’ GEORGE E.TARR*'~ ANDJOHN W.CRABB-~~ *Department of Biological Chemistry, University of Michigan, Ann Arbor, Michigan 48109, and lDepartment of Ophthalmology, University of Washington, Seattle, Washington 98195 Received January 31, 1983 Reverse-phase high-performance liquid chromatography (HPLC) resolution and recovery of cytochrome P-450 and bovine rhodopsin, both integral membrane proteins, and large peptides derived from P-450 LMI were enhanced by utilizing ternary solvents. Surprisingly, most test materials eluted later in the gradient when using mixtures of acetonitrile and propanol in the mobile phase compared to using either solvent alone. Of the supports tested, the best recovery of hydrophobic cytochrome P-450 LMI was experienced on the less retentive CN-bonded phase. Two alternate solvents for HPLC of polypeptides are proposed: (1) 0.02-o. 1 M hexafluoroacetone/ NH3, pH 7.2 for highly acidic peptides; and (2) 6 M formic acid/O.13 M trimethylamine, pH 1.5, vs 4 M formic acid/O.09 M trimethylamine in propanol for relatively insoluble peptides. Anomalous side reactions between formic acid and peptides can cause HPLC peak broadening, increased retention, and decreased resolution. These deleterious effects are thought to be due in part to formyl esterilication of serine and threonine residues and appear to be reversible by aminoethanol treatment. High-performance liquid chromatography, tides (3-7) and hydrophobic, membrane-as- particularly reverse-phase chromatography, sociated polypeptides (8- 14), methods for such has supplanted the more classical methods of fractionations are not yet well established. -

Cyanogen Bromide; CASRN 506-68-3
Integrated Risk Information System (IRIS) U.S. Environmental Protection Agency Chemical Assessment Summary National Center for Environmental Assessment Cyanogen bromide; CASRN 506-68-3 Human health assessment information on a chemical substance is included in the IRIS database only after a comprehensive review of toxicity data, as outlined in the IRIS assessment development process. Sections I (Health Hazard Assessments for Noncarcinogenic Effects) and II (Carcinogenicity Assessment for Lifetime Exposure) present the conclusions that were reached during the assessment development process. Supporting information and explanations of the methods used to derive the values given in IRIS are provided in the guidance documents located on the IRIS website. STATUS OF DATA FOR Cyanogen bromide File First On-Line 09/26/1988 Category (section) Assessment Available? Last Revised Oral RfD (I.A.) yes 09/26/1988* Inhalation RfC (I.B.) not evaluated Carcinogenicity Assessment (II.) not evaluated *A comprehensive review of toxicological studies was completed (2004) - please see section I.A.6 for more information. I. Chronic Health Hazard Assessments for Noncarcinogenic Effects I.A. Reference Dose for Chronic Oral Exposure (RfD) Substance Name — Cyanogen bromide CASRN — 506-68-3 Last Revised — 09/26/1988 The oral Reference Dose (RfD) is based on the assumption that thresholds exist for certain toxic effects such as cellular necrosis. It is expressed in units of mg/kg-day. In general, the RfD is an estimate (with uncertainty spanning perhaps an order of magnitude) of a daily exposure to the human population (including sensitive subgroups) that is likely to be without an appreciable risk 1 Integrated Risk Information System (IRIS) U.S. -
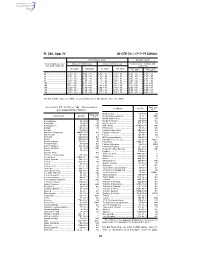
40 CFR Ch. I (7–1–19 Edition)
Pt. 266, App. IV 40 CFR Ch. I (7–1–19 Edition) Noncomplex terrain Complex terrain Terrain-adjusted effec- Values for urban areas Values for rural areas Values for use in urban and tive stack height (m) rural areas Cl2 (g/hr) HCl (g/hr) Cl2 (g/hr) HCl (g/hr) Cl2 (g/hr) HCl (g/hr) 80 .................................. 2.9E + 03 ....... 5.0E + 04 ....... 1.0E + 04 ....... 1.8E + 05 ....... 1.3E + 03 ....... 2.3E + 04 85 .................................. 3.3E + 03 ....... 5.8E + 04 ....... 1.2E + 04 ....... 2.2E + 05 ....... 1.4E + 03 ....... 2.5E + 04 90 .................................. 3.7E + 03 ....... 6.6E + 04 ....... 1.4E + 04 ....... 2.5E + 05 ....... 1.6E + 03 ....... 2.9E + 04 95 .................................. 4.2E + 03 ....... 7.4E + 04 ....... 1.7E + 04 ....... 3.0E + 05 ....... 1.8E + 03 ....... 3.2E + 04 100 ................................ 4.8E + 03 ....... 8.4E + 04 ....... 2.1E + 04 ....... 3.6E + 05 ....... 2.0E + 03 ....... 3.5E + 04 105 ................................ 5.3E + 03 ....... 9.2E + 04 ....... 2.4E + 04 ....... 4.3E + 05 ....... 2.3E + 03 ....... 3.9E + 04 110 ................................ 6.2E + 03 ....... 1.1E + 05 ....... 2.9E + 04 ....... 5.1E + 05 ....... 2.5E + 03 ....... 4.5E + 04 115 ................................ 7.2E + 03 ....... 1.3E + 05 ....... 3.5E + 04 ....... 6.1E + 05 ....... 2.8E + 03 ....... 5.0E + 04 120 ................................ 8.2E + 03 ....... 1.4E + 05 ....... 4.1E + 04 ....... 7.2E + 05 ....... 3.2E + 03 ....... 5.6E + 04 [56 FR 32691, July 17, 1991, as amended at 71 FR 40277, July 14, 2006] APPENDIX IV TO PART 266—REFERENCE RAC (ug/ Constituent CAS No. 3 AIR CONCENTRATIONS* m ) Methoxychlor ............................... 72–43–5 50 Constituent CAS No. -

Chemical Waste Disposal Guidance.Pdf
SOP Title: S.A.A. Generated Waste-(laboratories) SOP Effective Date: 02/01/2019 Page 1 of 54 Site: ☒ 263 Farmington Campus ☒ 400 Farmington Avenue ☒ 21 South Road EH&S Primary Content Approval Authority: Steven Jacobs, Director of EH&S EH&S Secondary Content Approval Authority: Robert Gottlieb, EH&S Specialist EH&S Document Control Administrator: Elizabeth Pokorski, Administrative Officer 1. PURPOSE & SCOPE Purpose- To define generation of regulated wastes, & the safe management procedures for these wastes at UConn Health’s Laboratory Satellite Accumulation Areas (S.A.A.’s). Scope- Define roles and responsibilities, basic rules & regulations regarding S.A.A. waste management; incorporating emergency contingency planning application & Personal Protective Equipment (PPE). 2.0 LISTED HAZARDOUS WASTES . Wastes that are specifically identified in one of four lists developed by U.S. EPA in the federal hazardous waste regulations [40 CFR 261.31 through 261.33]. Each hazardous waste listing includes a description of a specific type of waste that EPA considers hazardous enough to warrant regulation. The four groups of listed hazardous wastes are easily identified by the letter that begins their 4-digit EPA waste code (i.e., “F,” “K,” “U,” or “P”). See Appendix A, starting on page 13, for “LISTED HAZARDOUS WASTE CATAGORIES PER EPA.” 3.0 CHARACTERISTIC HAZARDOUS WASTE Subpart C—Characteristics of Hazardous Waste §261.20 General. [Comment: §262.11 of this chapter sets forth the generator's responsibility to determine whether his waste exhibits one or more of the characteristics identified in this subpart] (a) A solid waste, as defined in §261.2, which is not excluded from regulation as a hazardous waste under §261.4(b), is a hazardous waste if it exhibits any of the characteristics identified in this subpart. -

Medical Management of Chemical Casualties Handbook
US Army Medical Research Institute of Chemical Defense (USAMRICD) MEDICAL MANAGEMENT OF CHEMICAL CASUALTIES HANDBOOK Chemical Casualty Care Division (MCMR-UV-ZM) USAMRICD 3100 Ricketts Point Road Aberdeen Proving Ground, MD 21010-5400 THIRD EDITION July 2000 Disclaimer The purpose of this Handbook is to provide concise supplemental reading material for attendees of the Medical Management of Chemical Casualties Course. Every effort has been made to make the information contained in this Handbook consistent with official policy and doctrine. This Handbook, however, is not an official Department of the Army publication, nor is it official doctrine. It should not be construed as such unless it is supported by other documents. ii Table of Contents Introduction ................................................................................................................... 1 Pulmonary Agents....................................................................................................... 10 Cyanide ........................................................................................................................ 19 Table. Cyanide (AC and AK). Effects From Vapor Exposure ............................. 25 Vesicants ..................................................................................................................... 30 Mustard..................................................................................................................... 31 Table. Effects of Mustard Vapor .................................................................................... -

Cyanogen Agents
J R Army Med Corps 2002; 148: 383-386 J R Army Med Corps: first published as 10.1136/jramc-148-04-08 on 1 December 2002. Downloaded from Cyanogen Agents GENERAL HCN + Na2S2O3 ‘HSCN + Na2SO3 This reaction with sodium thiosulphate Introduction forms the basis of a therapy of cyanide Cyanogen agents produce their effects by poisoning. Metal ions easily form complexes interfering with oxygen utilization at the with cyanide ions for example: cellular level. Their toxicity is primarily derived from the liberation of the CN-group. - 2- - Inhalation is the usual route of entry. The CoCl2 + 4CN ‘Co(CN) 4+ 2Cl . term "blood agents" has, in the past been These types of reaction are employed in used to describe cyanogen agents and is still reactive carbons where the charcoal is in widespread use. It should be noted, impregnated with metal ions in order to however, that not all "blood agents" are increase the absorptive capacity of charcoal. cyanogens (e.g. carbon monoxide). Similar reactions with complexed metal ions The commodity industrial chemical are also utilised in some forms of therapy. hydrogen cyanide (HCN (AC)) was used during WWI and is still attractive as an Detection improvised weapons fill because of its Automatic detectors are available which availability. Hydrogen cyanide is released detect attack concentrations of cyanide when cyanides (e.g. acetone cyanohydrine, vapour. DraegerTM tubes are also available, as sodium cyanide, potassium cyanide) are are water testing kits. spilled in water or subjected to acid. The cyanogen halides, cyanogen chloride (ClCN Protection (CK)) and cyanogen bromide (BrCN), were Hydrogen cyanide, because of its volatility used during WWI. -

Cyanogen Bromide
Cyanogen bromide sc-203011 Material Safety Data Sheet Hazard Alert Code Key: EXTREME HIGH MODERATE LOW Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME Cyanogen bromide STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY0 HEALTH4 HAZARD INSTABILITY1 SUPPLIER Company: Santa Cruz Biotechnology, Inc. Address: 2145 Delaware Ave Santa Cruz, CA 95060 Telephone: 800.457.3801 or 831.457.3800 Emergency Tel: CHEMWATCH: From within the US and Canada: 877-715-9305 Emergency Tel: From outside the US and Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 PRODUCT USE Organic synthesis, parasiticide, fumigating compositions, rat exterminants, cyaniding reagent in gold extraction process. For selective peptide cleavage, e.g. methionine, and for use in protein immobilisation procedures. SYNONYMS Br-C-N, BrCN, "bromine cyanide", bromocyan, bromocyanide, bromocyanogen, campilit, cyanobromide, "cyanogen monobromide" Section 2 - HAZARDS IDENTIFICATION CANADIAN WHMIS SYMBOLS EMERGENCY OVERVIEW RISK Contact with acids liberates very toxic gas. Causes burns. Risk of serious damage to eyes. Very toxic by inhalation, in contact with skin and if swallowed. May cause long-term adverse effects in the environment. 1 of 23 Cyanogen bromide sc-203011 Material Safety Data Sheet Hazard Alert Code Key: EXTREME HIGH MODERATE LOW Very toxic to aquatic organisms, may cause long-term adverse effects in the aquatic environment. POTENTIAL HEALTH EFFECTS ACUTE HEALTH EFFECTS SWALLOWED ! Severely toxic effects may result from the accidental ingestion of the material; animal experiments indicate that ingestion of less than 5 gram may be fatal or may produce serious damage to the health of the individual. -

Provisional Peer-Reviewed Toxicity Values for Cyanogen Bromide (Casrn 506-68-3)
EPA/690/R-12/009F l Final 12-12-2012 Provisional Peer-Reviewed Toxicity Values for Cyanogen Bromide (CASRN 506-68-3) Superfund Health Risk Technical Support Center National Center for Environmental Assessment Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 AUTHORS, CONTRIBUTORS, AND REVIEWERS CHEMICAL MANAGER Harlal Choudhury, DVM, PhD, DABT National Center for Environmental Assessment, Cincinnati, OH DRAFT DOCUMENT PREPARED BY ICF International 9300 Lee Highway Fairfax, VA 22031 PRIMARY INTERNAL REVIEWERS Ghazi Dannan, PhD National Center for Environmental Assessment, Washington, DC Anuradha Mudipalli, MSc, PhD National Center for Environmental Assessment, Research Triangle Park, NC This document was externally peer reviewed under contract to Eastern Research Group, Inc. 110 Hartwell Avenue Lexington, MA 02421-3136 Questions regarding the contents of this document may be directed to the U.S. EPA Office of Research and Development’s National Center for Environmental Assessment, Superfund Health Risk Technical Support Center (513-569-7300). i Cyanogen Bromide TABLE OF CONTENTS COMMONLY USED ABBREVIATIONS ................................................................................... iii BACKGROUND .............................................................................................................................1 DISCLAIMERS ...............................................................................................................................1 QUESTIONS REGARDING PPRTVs -

Epa Hazardous Waste Codes
EPA HAZARDOUS WASTE CODES Code Waste description Code Waste description D001 Ignitable waste D023 o-Cresol D002 Corrosive waste D024 m-Cresol D003 Reactive waste D025 p-Cresol D004 Arsenic D026 Cresol D005 Barium D027 1,4-Dichlorobenzene D006 Cadmium D028 1,2-Dichloroethane D007 Chromium D029 1,1-Dichloroethylene D008 Lead D030 2,4-Dinitrotoluene D009 Mercury D031 Heptachlor (and its epoxide) D010 Selenium D032 Hexachlorobenzene D011 Silver D033 Hexachlorobutadiene D012 Endrin(1,2,3,4,10,10-hexachloro-1,7-epoxy- D034 Hexachloroethane 1,4,4a,5,6,7,8,8a-octahydro-1,4-endo, endo- 5,8-dimeth-ano-naphthalene) D035 Methyl ethyl ketone D013 Lindane (1,2,3,4,5,6-hexa- D036 Nitrobenzene chlorocyclohexane, gamma isomer) D037 Pentachlorophenol D014 Methoxychlor (1,1,1-trichloro-2,2-bis [p- methoxyphenyl] ethane) D038 Pyridine D015 Toxaphene (C10 H10 Cl8, Technical D039 Tetrachloroethylene chlorinated camphene, 67-69 percent chlorine) D040 Trichlorethylene D016 2,4-D (2,4-Dichlorophenoxyacetic acid) D041 2,4,5-Trichlorophenol D017 2,4,5-TP Silvex (2,4,5- D042 2,4,6-Trichlorophenol Trichlorophenoxypropionic acid) D043 Vinyl chloride D018 Benzene D019 Carbon tetrachloride D020 Chlordane D021 Chlorobenzene D022 Chloroform B-1 EPA HAZARDOUS WASTE CODES (Continued) Code Waste description Code Waste description HAZARDOUS WASTE FROM NONSPECIFIC solvents: cresols, cresylic acid, and SOURCES nitrobenzene; and the still bottoms from the recovery of these solvents; all spent solvent F001 The following spent halogenated solvents mixtures/blends containing, before use, a used in degreasing: Tetrachloroethylene, total of ten percent or more (by volume) of trichlorethylene, methylene chloride, 1,1,1- one or more of the above nonhalogenated trichloroethane, carbon tetrachloride and solvents or those solvents listed in F001, chlorinated fluorocarbons; all spent solvent F002, and F005; and still bottoms from the mixtures/blends used in degreasing recovery of these spent solvents and spent containing, before use, a total of ten solvent mixtures. -

Immobilization
IMMOBILIZATION Prof. Alejandro Hochkoeppler Department of Pharmaceutical Sciences and Biotechnology University of Bologna E-mail: [email protected] IMMOBILIZATION Advantages of enzyme immobilization: • Enzyme can be recycled Enzymes are expensive! • Improved stability Disadvantages of enzyme immobilization: • Lower specific activity (harsh conditions) Stability/inactivation • Accessibility of active site Methods of immobilization: • adsorbtion (insoluble particles, membranes…). Mild conditions. • entrapment (gels, microspheres, cartridges). Mild conditions. • covalent linkage (nylon, glass, resins…): Harsh conditions. IMMOBILIZATION Immobilization by entrapment. Enzyme-containing fibers. Dinelli et al., 1976, Methods in Enzymology, 44: 227-243 • A polymer suitable for fiber production is dispersed in organic solvent (water-immiscible) • Addition of aqueous enzyme solution, emulsion preparation • Transfer of the emulsion to a solvent able to trigger coagulation (formation of fibers) • The pores of fibers let the substrate (product) diffuse in (out) • Enzyme is confined to the internal compartment of fibers and cannot diffuse out • Reaction rate can be affected by diffusion of substrate and/or product • The enzyme is subjected to a mild treatment: retention of activity. • The amount of enzyme entrapped depends on the stock enzyme concentration and on the water content attainable by the emulsion. IMMOBILIZATION Preparation of enzyme-containing fibers P P diameter: 0.08-0.125 µm Emulsion Roller D2: reel of fibers IMMOBILIZATION Preparation