Polymer Solutions: an Introduction to Physical Properties
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modelling and Numerical Simulation of Phase Separation in Polymer Modified Bitumen by Phase- Field Method
http://www.diva-portal.org Postprint This is the accepted version of a paper published in Materials & design. This paper has been peer- reviewed but does not include the final publisher proof-corrections or journal pagination. Citation for the original published paper (version of record): Zhu, J., Lu, X., Balieu, R., Kringos, N. (2016) Modelling and numerical simulation of phase separation in polymer modified bitumen by phase- field method. Materials & design, 107: 322-332 http://dx.doi.org/10.1016/j.matdes.2016.06.041 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:kth:diva-188830 ACCEPTED MANUSCRIPT Modelling and numerical simulation of phase separation in polymer modified bitumen by phase-field method Jiqing Zhu a,*, Xiaohu Lu b, Romain Balieu a, Niki Kringos a a Department of Civil and Architectural Engineering, KTH Royal Institute of Technology, Brinellvägen 23, SE-100 44 Stockholm, Sweden b Nynas AB, SE-149 82 Nynäshamn, Sweden * Corresponding author. Email: [email protected] (J. Zhu) Abstract In this paper, a phase-field model with viscoelastic effects is developed for polymer modified bitumen (PMB) with the aim to describe and predict the PMB storage stability and phase separation behaviour. The viscoelastic effects due to dynamic asymmetry between bitumen and polymer are represented in the model by introducing a composition-dependent mobility coefficient. A double-well potential for PMB system is proposed on the basis of the Flory-Huggins free energy of mixing, with some simplifying assumptions made to take into account the complex chemical composition of bitumen. -

Solutes and Solution
Solutes and Solution The first rule of solubility is “likes dissolve likes” Polar or ionic substances are soluble in polar solvents Non-polar substances are soluble in non- polar solvents Solutes and Solution There must be a reason why a substance is soluble in a solvent: either the solution process lowers the overall enthalpy of the system (Hrxn < 0) Or the solution process increases the overall entropy of the system (Srxn > 0) Entropy is a measure of the amount of disorder in a system—entropy must increase for any spontaneous change 1 Solutes and Solution The forces that drive the dissolution of a solute usually involve both enthalpy and entropy terms Hsoln < 0 for most species The creation of a solution takes a more ordered system (solid phase or pure liquid phase) and makes more disordered system (solute molecules are more randomly distributed throughout the solution) Saturation and Equilibrium If we have enough solute available, a solution can become saturated—the point when no more solute may be accepted into the solvent Saturation indicates an equilibrium between the pure solute and solvent and the solution solute + solvent solution KC 2 Saturation and Equilibrium solute + solvent solution KC The magnitude of KC indicates how soluble a solute is in that particular solvent If KC is large, the solute is very soluble If KC is small, the solute is only slightly soluble Saturation and Equilibrium Examples: + - NaCl(s) + H2O(l) Na (aq) + Cl (aq) KC = 37.3 A saturated solution of NaCl has a [Na+] = 6.11 M and [Cl-] = -

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Chapter 2: Basic Tools of Analytical Chemistry
Chapter 2 Basic Tools of Analytical Chemistry Chapter Overview 2A Measurements in Analytical Chemistry 2B Concentration 2C Stoichiometric Calculations 2D Basic Equipment 2E Preparing Solutions 2F Spreadsheets and Computational Software 2G The Laboratory Notebook 2H Key Terms 2I Chapter Summary 2J Problems 2K Solutions to Practice Exercises In the chapters that follow we will explore many aspects of analytical chemistry. In the process we will consider important questions such as “How do we treat experimental data?”, “How do we ensure that our results are accurate?”, “How do we obtain a representative sample?”, and “How do we select an appropriate analytical technique?” Before we look more closely at these and other questions, we will first review some basic tools of importance to analytical chemists. 13 14 Analytical Chemistry 2.0 2A Measurements in Analytical Chemistry Analytical chemistry is a quantitative science. Whether determining the concentration of a species, evaluating an equilibrium constant, measuring a reaction rate, or drawing a correlation between a compound’s structure and its reactivity, analytical chemists engage in “measuring important chemical things.”1 In this section we briefly review the use of units and significant figures in analytical chemistry. 2A.1 Units of Measurement Some measurements, such as absorbance, A measurement usually consists of a unit and a number expressing the do not have units. Because the meaning of quantity of that unit. We may express the same physical measurement with a unitless number may be unclear, some authors include an artificial unit. It is not different units, which can create confusion. For example, the mass of a unusual to see the abbreviation AU, which sample weighing 1.5 g also may be written as 0.0033 lb or 0.053 oz. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Properties of Two-Dimensional Polymers
Macromolecules 1982,15, 549-553 549 The squared frequencies uA2are obtained from the ei- (4) Flory, P. J. "Statistical Mechanics of Chain Molecules"; In- genvalues Of this matrix described in the text* terscience: New York, 1969; p 315. (5) Volkenstein,M. V. &Configurational Statistics of polymeric for N = 3 and 4 will be found in ref 7. Chains" (translated from the Russian edition by S. N. Ti- masheff and M. J. Timasheff);Interscience: New York, 1963; References and Notes p 450. (1) Weiner, J. H.; Perchak, D. Macromolecules 1981, 14, 1590. (6) Frenkel, J. "Kinetic Theory of Liquids"; Oxford University (2) Weiner, J. H., Macromolecules, preceding paper in this issue. Press: London, 1946, Dover reprint, 1955; pp 474-476. (3) G6, N.; Scheraga, H. A. Macromolecules 1976, 9, 535. (7) Perchak, D. Doctoral Dissertation, Brown University, 1981. Properties of Two-Dimensional Polymers Jan Tobochnik and Itzhak Webman* Department of Mathematics, Rutgers, The State University, New Brunswick, New Jersey 08903 Joel L. Lebowitzt Institute for Advanced Study, Princeton, New Jersey 08540 M. H. Kalos Courant Institute of Mathematical Sciences, New York university, New York, New York 10012. Received August 3, 1981 ABSTRACT: We have performed a series of Monte Carlo simulations for a two-dimensional polymer chain with monomers interacting via a Lennard-Jones potential. An analysis of a mean field theory, based on approximating the free energy as the sum of an elastic part and a fluid part, shows that in two dimensions there is a sharp collapse transition, at T = 0, but that there is no ideal or quasi-ideal behavior at the transition as there is in three dimensions. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Chapter 15: Solutions
452-487_Ch15-866418 5/10/06 10:51 AM Page 452 CHAPTER 15 Solutions Chemistry 6.b, 6.c, 6.d, 6.e, 7.b I&E 1.a, 1.b, 1.c, 1.d, 1.j, 1.m What You’ll Learn ▲ You will describe and cate- gorize solutions. ▲ You will calculate concen- trations of solutions. ▲ You will analyze the colliga- tive properties of solutions. ▲ You will compare and con- trast heterogeneous mixtures. Why It’s Important The air you breathe, the fluids in your body, and some of the foods you ingest are solu- tions. Because solutions are so common, learning about their behavior is fundamental to understanding chemistry. Visit the Chemistry Web site at chemistrymc.com to find links about solutions. Though it isn’t apparent, there are at least three different solu- tions in this photo; the air, the lake in the foreground, and the steel used in the construction of the buildings are all solutions. 452 Chapter 15 452-487_Ch15-866418 5/10/06 10:52 AM Page 453 DISCOVERY LAB Solution Formation Chemistry 6.b, 7.b I&E 1.d he intermolecular forces among dissolving particles and the Tattractive forces between solute and solvent particles result in an overall energy change. Can this change be observed? Safety Precautions Dispose of solutions by flushing them down a drain with excess water. Procedure 1. Measure 10 g of ammonium chloride (NH4Cl) and place it in a Materials 100-mL beaker. balance 2. Add 30 mL of water to the NH4Cl, stirring with your stirring rod. -

Lecture 3. the Basic Properties of the Natural Atmosphere 1. Composition
Lecture 3. The basic properties of the natural atmosphere Objectives: 1. Composition of air. 2. Pressure. 3. Temperature. 4. Density. 5. Concentration. Mole. Mixing ratio. 6. Gas laws. 7. Dry air and moist air. Readings: Turco: p.11-27, 38-43, 366-367, 490-492; Brimblecombe: p. 1-5 1. Composition of air. The word atmosphere derives from the Greek atmo (vapor) and spherios (sphere). The Earth’s atmosphere is a mixture of gases that we call air. Air usually contains a number of small particles (atmospheric aerosols), clouds of condensed water, and ice cloud. NOTE : The atmosphere is a thin veil of gases; if our planet were the size of an apple, its atmosphere would be thick as the apple peel. Some 80% of the mass of the atmosphere is within 10 km of the surface of the Earth, which has a diameter of about 12,742 km. The Earth’s atmosphere as a mixture of gases is characterized by pressure, temperature, and density which vary with altitude (will be discussed in Lecture 4). The atmosphere below about 100 km is called Homosphere. This part of the atmosphere consists of uniform mixtures of gases as illustrated in Table 3.1. 1 Table 3.1. The composition of air. Gases Fraction of air Constant gases Nitrogen, N2 78.08% Oxygen, O2 20.95% Argon, Ar 0.93% Neon, Ne 0.0018% Helium, He 0.0005% Krypton, Kr 0.00011% Xenon, Xe 0.000009% Variable gases Water vapor, H2O 4.0% (maximum, in the tropics) 0.00001% (minimum, at the South Pole) Carbon dioxide, CO2 0.0365% (increasing ~0.4% per year) Methane, CH4 ~0.00018% (increases due to agriculture) Hydrogen, H2 ~0.00006% Nitrous oxide, N2O ~0.00003% Carbon monoxide, CO ~0.000009% Ozone, O3 ~0.000001% - 0.0004% Fluorocarbon 12, CF2Cl2 ~0.00000005% Other gases 1% Oxygen 21% Nitrogen 78% 2 • Some gases in Table 3.1 are called constant gases because the ratio of the number of molecules for each gas and the total number of molecules of air do not change substantially from time to time or place to place. -
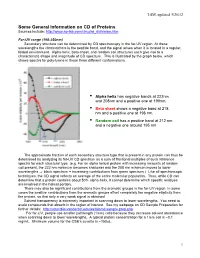
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Lecture Notes on Structure and Properties of Engineering Polymers
Structure and Properties of Engineering Polymers Lecture: Microstructures in Polymers Nikolai V. Priezjev Textbook: Plastics: Materials and Processing (Third Edition), by A. Brent Young (Pearson, NJ, 2006). Microstructures in Polymers • Gas, liquid, and solid phases, crystalline vs. amorphous structure, viscosity • Thermal expansion and heat distortion temperature • Glass transition temperature, melting temperature, crystallization • Polymer degradation, aging phenomena • Molecular weight distribution, polydispersity index, degree of polymerization • Effects of molecular weight, dispersity, branching on mechanical properties • Melt index, shape (steric) effects Reading: Chapter 3 of Plastics: Materials and Processing by A. Brent Strong https://www.slideshare.net/NikolaiPriezjev Gas, Liquid and Solid Phases At room temperature Increasing density Solid or liquid? Pitch Drop Experiment Pitch (derivative of tar) at room T feels like solid and can be shattered by a hammer. But, the longest experiment shows that it flows! In 1927, Professor Parnell at UQ heated a sample of pitch and poured it into a glass funnel with a sealed stem. Three years were allowed for the pitch to settle, and in 1930 the sealed stem was cut. From that date on the pitch has slowly dripped out of the funnel, with seven drops falling between 1930 and 1988, at an average of one drop every eight years. However, the eight drop in 2000 and the ninth drop in 2014 both took about 13 years to fall. It turns out to be about 100 billion times more viscous than water! Pitch, before and after being hit with a hammer. http://smp.uq.edu.au/content/pitch-drop-experiment Liquid phases: polymer melt vs.