Coordinated Activation of the RAC-GAP Β2-CHIMAERIN by an Atypical Proline-Rich Domain and Diacylglcercol Bruce J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
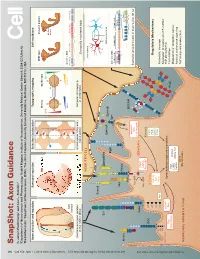
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

The Proximal Signaling Network of the BCR-ABL1 Oncogene Shows a Modular Organization
Oncogene (2010) 29, 5895–5910 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 www.nature.com/onc ORIGINAL ARTICLE The proximal signaling network of the BCR-ABL1 oncogene shows a modular organization B Titz, T Low, E Komisopoulou, SS Chen, L Rubbi and TG Graeber Crump Institute for Molecular Imaging, Institute for Molecular Medicine, Jonsson Comprehensive Cancer Center, California NanoSystems Institute, Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA, USA BCR-ABL1 is a fusion tyrosine kinase, which causes signaling effects of BCR-ABL1 toward leukemic multiple types of leukemia. We used an integrated transformation. proteomic approach that includes label-free quantitative Oncogene (2010) 29, 5895–5910; doi:10.1038/onc.2010.331; protein complex and phosphorylation profiling by mass published online 9 August 2010 spectrometry to systematically characterize the proximal signaling network of this oncogenic kinase. The proximal Keywords: adaptor protein; BCR-ABL1; phospho- BCR-ABL1 signaling network shows a modular and complex; quantitative mass spectrometry; signaling layered organization with an inner core of three leukemia network; systems biology transformation-relevant adaptor protein complexes (Grb2/Gab2/Shc1 complex, CrkI complex and Dok1/ Dok2 complex). We introduced an ‘interaction direction- ality’ analysis, which annotates static protein networks Introduction with information on the directionality of phosphorylation- dependent interactions. In this analysis, the observed BCR-ABL1 is a constitutively active oncogenic fusion network structure was consistent with a step-wise kinase that arises through a chromosomal translocation phosphorylation-dependent assembly of the Grb2/Gab2/ and causes multiple types of leukemia. It is found in Shc1 and the Dok1/Dok2 complexes on the BCR-ABL1 many cases (B25%) of adult acute lymphoblastic core. -

Mutation-Specific and Common Phosphotyrosine Signatures of KRAS G12D and G13D Alleles Anticipated Graduation August 1St, 2018
MUTATION-SPECIFIC AND COMMON PHOSPHOTYROSINE SIGNATURES OF KRAS G12D AND G13D ALLELES by Raiha Tahir A dissertation submitted to The Johns Hopkins University in conformity with the requirement of the degree of Doctor of Philosophy Baltimore, MD August 2018 © 2018 Raiha Tahir All Rights Reserved ABSTRACT KRAS is one of the most frequently mutated genes across all cancer subtypes. Two of the most frequent oncogenic KRAS mutations observed in patients result in glycine to aspartic acid substitution at either codon 12 (G12D) or 13 (G13D). Although the biochemical differences between these two predominant mutations are not fully understood, distinct clinical features of the resulting tumors suggest involvement of disparate signaling mechanisms. When we compared the global phosphotyrosine proteomic profiles of isogenic colorectal cancer cell lines bearing either G12D or G13D KRAS mutations, we observed both shared as well as unique signaling events induced by the two KRAS mutations. Remarkably, while the G12D mutation led to an increase in membrane proximal and adherens junction signaling, the G13D mutation led to activation of signaling molecules such as non-receptor tyrosine kinases, MAPK kinases and regulators of metabolic processes. The importance of one of the cell surface molecules, MPZL1, which found to be hyperphosphorylated in G12D cells, was confirmed by cellular assays as its knockdown led to a decrease in proliferation of G12D but not G13D expressing cells. Overall, our study reveals important signaling differences across two common KRAS mutations and highlights the utility of our approach to systematically dissect the subtle differences between related oncogenic mutants and potentially lead to individualized treatments. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

CD93 and Dystroglycan Cooperation in Human Endothelial Cell Adhesion and Migration
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 9 CD93 and dystroglycan cooperation in human endothelial cell adhesion and migration Federico Galvagni1,*, Federica Nardi1,*, Marco Maida1, Giulia Bernardini1, Silvia Vannuccini2, Felice Petraglia2, Annalisa Santucci1, Maurizio Orlandini1 1 Department of Biotechnology, Chemistry and Pharmacy, University of Siena, 2-53100 Siena, Italy 2 Department of Molecular and Developmental Medicine, Obstetrics and Gynecology, University of Siena, 53100 Siena, Italy *These authors contributed equally to this work Correspondence to: Maurizio Orlandini, e-mail: [email protected] Keywords: angiogenesis, signal transduction, C1qRp, Src, Cbl Received: June 29, 2015 Accepted: January 22, 2016 Published: February 02, 2016 ABSTRACT CD93 is a transmembrane glycoprotein predominantly expressed in endothelial cells. Although CD93 displays proangiogenic activity, its molecular function in angiogenesis still needs to be clarified. To get molecular insight into the biological role of CD93 in the endothelium, we performed proteomic analyses to examine changes in the protein profile of endothelial cells after CD93 silencing. Among differentially expressed proteins, we identified dystroglycan, a laminin-binding protein involved in angiogenesis, whose expression is increased in vascular endothelial cells within malignant tumors. Using immunofluorescence, FRET, and proximity ligation analyses, we observed a close interaction between CD93 and β-dystroglycan. Moreover, silencing experiments showed that CD93 and dystroglycan promoted endothelial cell migration and organization into capillary-like structures. CD93 proved to be phosphorylated on tyrosine 628 and 644 following cell adhesion on laminin through dystroglycan. This phosphorylation was shown to be necessary for a proper endothelial migratory phenotype. Moreover, we showed that during cell spreading phosphorylated CD93 recruited the signaling protein Cbl, which in turn was phosphorylated on tyrosine 774. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Expression and Localization of Grass Carp Pkc-Θ (Protein Kinase C Theta) Gene After Its Activation T
Fish and Shellfish Immunology 87 (2019) 788–795 Contents lists available at ScienceDirect Fish and Shellfish Immunology journal homepage: www.elsevier.com/locate/fsi Full length article Expression and localization of grass carp pkc-θ (protein kinase C theta) gene after its activation T ∗∗ Rumana Mehjabina,b,1, Liangming Chena,b,1, Rong Huanga, , Denghui Zhua,b, Cheng Yanga,b, ∗ Yongming Lia, Lanjie Liaoa, Libo Hea, Zuoyan Zhua, Yaping Wanga, a State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, 430072, China b University of Chinese Academy of Sciences, Beijing, 100049, China ARTICLE INFO ABSTRACT Keywords: Haemorrhagic disease caused by grass carp reovirus (GCRV) can result in large-scale death of young grass carp, Grass carp leading to irreparable economic losses that seriously affect large-scale breeding. Protein kinase C (PKC, also Grass carp reovirus (GCRV) known as PRKC) represents a family of serine/threonine protein kinases that includes multiple isozymes in many Protein kinase C (PKC) species. Among these, PKC-θ (PKC theta, also written as PRKCQ) is a novel isoform, mainly expressed in T cells, Host immune defences that is known to be involved in immune system function in mammals. To date, no research on immunological functions of fish Pkc-θ has been reported. To address this issue, we cloned the grass carp pkc-θ gene. Phylogenetic and syntenic analysis showed that this gene is the most evolutionarily conserved relative to zeb- rafish. Real-time quantitative PCR (RT-qPCR) indicated that pkc-θ was expressed at high levels in the gills and spleen of healthy grass carp. -

Supplementary Materials
Supplementary material. S1. Images from automatic imaging reader Cytation™ 1. A, A’, A’’ present the same field of view. Cells migrating from the upper compartment of the inserts are stained with DID (red color), Cell nuclei are stained with DAPI (blue color). A’ and A’’ demonstrate the method of analysis (A’ – nuclei numbering, A’’ – migrating cells numbering), scale bar – 300 µm. ASC BM-MSC Y BM-MSC A mean SEM mean SEM mean SEM CXCL6 2.240 0.727 4.158 0.677 2.149 1.816 CXCL16 4.277 0.248 0.763 0.405 1.130 0.346 CXCL12 -2.855 0.483 -4.528 0.226 -3.616 0.318 SMAD3 -0.511 0.191 -1.522 0.188 -1.332 0.215 TGFB2 3.742 0.533 1.238 0.210 0.990 0.568 COL14A 1.532 0.357 -0.397 0.736 -1.522 0.469 MHX 1.397 0.414 1.145 0.412 0.642 0.613 SCX 2.984 0.301 2.062 0.320 2.031 0.249 RUNX2 3.290 0.359 2.319 0.388 2.546 0.529 PPRAG 1.720 0.303 2.926 0.423 2.912 0.215 Supplementary material S3. The table provides the mean Δct and SEM values for all genes and all groups analyzed in this study (n=8 in each group). Supplementary material S2. The table provides the list of genes which displayed significantly different expression between hASCs and hBM-MSCs A based on microarray nr Exp p-value Exp Fold Change ID Symbol Entrez Gene Name 1 2.84E-05 16.896 16960355 TM4SF1 transmembrane 4 L six family member 1 2 9.10E-09 12.238 16684080 IFI6 interferon alpha inducible protein 6 3 3.93E-08 11.851 16667702 VCAM1 vascular cell adhesion molecule 1 4 1.54E-05 9.944 16840113 CXCL16 C-X-C motif chemokine ligand 16 5 2.01E-06 9.123 16852463 RAB27B RAB27B, member RAS oncogene -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

Identification of Two Signaling Submodules Within the Crkii/ELMO
Cell Death and Differentiation (2007) 14, 963–972 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells A-C Tosello-Trampont1,4, JM Kinchen1,2,4, E Brugnera1,3, LB Haney1, MO Hengartner2 and KS Ravichandran*,1 Removal of apoptotic cells is a dynamic process coordinated by ligands on apoptotic cells, and receptors and other signaling proteins on the phagocyte. One of the fundamental challenges is to understand how different phagocyte proteins form specific and functional complexes to orchestrate the recognition/removal of apoptotic cells. One evolutionarily conserved pathway involves the proteins cell death abnormal (CED)-2/chicken tumor virus no. 10 (CT10) regulator of kinase (Crk)II, CED-5/180 kDa protein downstream of chicken tumor virus no. 10 (Crk) (Dock180), CED-12/engulfment and migration (ELMO) and MIG-2/RhoG, leading to activation of the small GTPase CED-10/Rac and cytoskeletal remodeling to promote corpse uptake. Although the role of ELMO : Dock180 in regulating Rac activation has been well defined, the function of CED-2/CrkII in this complex is less well understood. Here, using functional studies in cell lines, we observe that a direct interaction between CrkII and Dock180 is not required for efficient removal of apoptotic cells. Similarly, mutants of CED-5 lacking the CED-2 interaction motifs could rescue engulfment and migration defects in CED-5 deficient worms. Mutants of CrkII and Dock180 that could not biochemically interact could colocalize in membrane ruffles.