Uttrykking Final Ph[1].D THESIS TUYEN 27.10.06
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Chain Gene Induced by GM-CSF Β Receptor Regulation of Human High-Affinity Ige Molecular Mechanisms for Transcriptional
Molecular Mechanisms for Transcriptional Regulation of Human High-Affinity IgE Receptor β-Chain Gene Induced by GM-CSF This information is current as Kyoko Takahashi, Natsuko Hayashi, Shuichi Kaminogawa of September 23, 2021. and Chisei Ra J Immunol 2006; 177:4605-4611; ; doi: 10.4049/jimmunol.177.7.4605 http://www.jimmunol.org/content/177/7/4605 Downloaded from References This article cites 39 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/177/7/4605.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 23, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Molecular Mechanisms for Transcriptional Regulation of Human High-Affinity IgE Receptor -Chain Gene Induced by GM-CSF1 Kyoko Takahashi,*† Natsuko Hayashi,*‡ Shuichi Kaminogawa,† and Chisei Ra2* The -chain of the high-affinity receptor for IgE (FcRI) plays an important role in regulating activation of FcRI-expressing cells such as mast cells in allergic reactions. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Figure S1. Basic Information of RNA-Seq Results. (A) Bar Plot of Reads Component for Each Sample
Figure S1. Basic information of RNA-seq results. (A) Bar plot of reads component for each sample. (B) Dot plot shows the principal component analysis (PCA) of each sample. (C) Venn diagram of DEGs for three time points, the overlap part of the circles represents common differentially expressed genes between combinations. Figure S2. Scatter plot of DEGs for each time point. The X and Y axes represent the logarithmic value of gene expression. Red represents up-regulated DEG, blue represents down-regulated DEG, and gray represents non-DEG. Table S1. Primers used for quantitative real-time PCR analysis of DEGs. Gene Primer Sequence Forward 5’-CTACGAGTGGATGGTCAAGAGC-3’ FOXO1 Reverse 5’-CCAGTTCCTTCATTCTGCACACG-3’ Forward 5’-GACGTCCGGCATCAGAGAAA-3’ IRS2 Reverse 5’-TCCACGGCTAATCGTCACAG-3’ Forward 5’-CACAACCAGGACCTCACACC-3’ IRS1 Reverse 5’-CTTGGCACGATAGAGAGCGT-3’ Forward 5’-AGGATACCACTCCCAACAGACCT-3’ IL6 Reverse 5’-CAAGTGCATCATCGTTGTTCATAC-3’ Forward 5’-TCACGTTGTACGCAGCTACC-3’ CCL5 Reverse 5’-CAGTCCTCTTACAGCCTTTGG-3’ Forward 5’-CTGTGCAGCCGCAGTGCCTACC-3’ BMP7 Reverse 5’-ATCCCTCCCCACCCCACCATCT-3’ Forward 5’-CTCTCCCCCTCGACTTCTGA-3’ BCL2 Reverse 5’-AGTCACGCGGAACACTTGAT-3’ Forward 5’-CTGTCGAACACAGTGGTACCTG-3’ FGF7 Reverse 5’-CCAACTGCCACTGTCCTGATTTC-3’ Forward 5’-GGGAGCCAAAAGGGTCATCA-3’ GAPDH Reverse 5’-CGTGGACTGTGGTCATGAGT-3’ Supplementary material: Differentially expressed genes log2(SADS-CoV_12h/ Qvalue (SADS-CoV _12h/ Gene Symbol Control_12h) Control_12h) PTGER4 -1.03693 6.79E-04 TMEM72 -3.08132 3.66E-04 IFIT2 -1.02918 2.11E-07 FRAT2 -1.09282 4.66E-05 -

UPF1: from Mrna Surveillance to Protein Quality Control
biomedicines Review UPF1: From mRNA Surveillance to Protein Quality Control Hyun Jung Hwang 1,2, Yeonkyoung Park 1,2 and Yoon Ki Kim 1,2,* 1 Creative Research Initiatives Center for Molecular Biology of Translation, Korea University, Seoul 02841, Korea; [email protected] (H.J.H.); [email protected] (Y.P.) 2 Division of Life Sciences, Korea University, Seoul 02841, Korea * Correspondence: [email protected] Abstract: Selective recognition and removal of faulty transcripts and misfolded polypeptides are crucial for cell viability. In eukaryotic cells, nonsense-mediated mRNA decay (NMD) constitutes an mRNA surveillance pathway for sensing and degrading aberrant transcripts harboring premature termination codons (PTCs). NMD functions also as a post-transcriptional gene regulatory mechanism by downregulating naturally occurring mRNAs. As NMD is activated only after a ribosome reaches a PTC, PTC-containing mRNAs inevitably produce truncated and potentially misfolded polypeptides as byproducts. To cope with the emergence of misfolded polypeptides, eukaryotic cells have evolved sophisticated mechanisms such as chaperone-mediated protein refolding, rapid degradation of misfolded polypeptides through the ubiquitin–proteasome system, and sequestration of misfolded polypeptides to the aggresome for autophagy-mediated degradation. In this review, we discuss how UPF1, a key NMD factor, contributes to the selective removal of faulty transcripts via NMD at the molecular level. We then highlight recent advances on UPF1-mediated communication between mRNA surveillance and protein quality control. Keywords: nonsense-mediated mRNA decay; UPF1; aggresome; CTIF; mRNA surveillance; protein quality control Citation: Hwang, H.J.; Park, Y.; Kim, Y.K. UPF1: From mRNA Surveillance to Protein Quality Control. Biomedicines 2021, 9, 995. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Structure of the Activated Edc1-Dcp1-Dcp2-Edc3 Mrna Decapping Complex with Substrate Analog Poised for Catalysis
ARTICLE DOI: 10.1038/s41467-018-03536-x OPEN Structure of the activated Edc1-Dcp1-Dcp2-Edc3 mRNA decapping complex with substrate analog poised for catalysis Jeffrey S. Mugridge1, Ryan W. Tibble1,2, Marcin Ziemniak3,4,5, Jacek Jemielity4 & John D. Gross 1,2 The conserved decapping enzyme Dcp2 recognizes and removes the 5′ eukaryotic cap from mRNA transcripts in a critical step of many cellular RNA decay pathways. Dcp2 is a dynamic 1234567890():,; enzyme that functions in concert with the essential activator Dcp1 and a diverse set of coactivators to selectively and efficiently decap target mRNAs in the cell. Here we present a 2.84 Å crystal structure of K. lactis Dcp1–Dcp2 in complex with coactivators Edc1 and Edc3, and with substrate analog bound to the Dcp2 active site. Our structure shows how Dcp2 recognizes cap substrate in the catalytically active conformation of the enzyme, and how coactivator Edc1 forms a three-way interface that bridges the domains of Dcp2 to consolidate the active conformation. Kinetic data reveal Dcp2 has selectivity for the first transcribed nucleotide during the catalytic step. The heterotetrameric Edc1–Dcp1–Dcp2–Edc3 structure shows how coactivators Edc1 and Edc3 can act simultaneously to activate decapping catalysis. 1 Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, CA 94158, USA. 2 Program in Chemistry and Chemical Biology, University of California, San Francisco, San Francisco, CA 94158, USA. 3 Division of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, 02-089 Warsaw, Poland. 4 Centre of New Technologies, University of Warsaw, 02-097 Warsaw, Poland. -

Control of Gene Expression Through the Nonsense-Mediated RNA Decay Pathway
Nickless et al. Cell Biosci (2017) 7:26 DOI 10.1186/s13578-017-0153-7 Cell & Bioscience REVIEW Open Access Control of gene expression through the nonsense‑mediated RNA decay pathway Andrew Nickless1†, Julie M. Bailis2† and Zhongsheng You1* Abstract Nonsense-mediated RNA decay (NMD) was originally discovered as a cellular surveillance pathway that safeguards the quality of mRNA transcripts in eukaryotic cells. In its canonical function, NMD prevents translation of mutant mRNAs harboring premature termination codons (PTCs) by targeting them for degradation. However, recent stud- ies have shown that NMD has a much broader role in gene expression by regulating the stability of many normal transcripts. In this review, we discuss the function of NMD in normal physiological processes, its dynamic regulation by developmental and environmental cues, and its association with human disease. Keywords: Nonsense-mediated decay, RNA surveillance, Gene expression, Human disease Background indirectly in certain cellular milieus [1, 10, 11]. Although Nonsense-mediated RNA decay (NMD) is an essential the overall process of NMD is conserved for both mutant RNA quality control and gene regulatory mechanism that and non-mutant transcripts, the signals that trigger is conserved among eukaryotes [1–9]. NMD safeguards NMD or its inhibition vary according to the specifc tar- the quality of the transcriptome and maintains cellular get and biological context. homeostasis by eliminating transcripts that harbor pre- In this review we focus on the function and impact of mature termination codons (PTCs). PTCs can arise from NMD on normal gene expression in mammals. We out- errors in nucleic acid metabolism, such as genetic muta- line the NMD process and highlight the known roles for tions or defects in splicing or transcription. -

Arp31946 P050
Aviva Systems Biology HNF4A Antibody - N-terminal region (ARP31946_P050) Product Number ARP31946_P050 Product Page http://www.avivasysbio.com/hnf4a-antibody-n-terminal-region-arp31946-p050.html Product Name HNF4A Antibody - N-terminal region (ARP31946_P050) Size 100 ul Gene Symbol HNF4A Alias Symbols HNF4, HNF4a7, HNF4a8, HNF4a9, HNF4alpha, MODY, MODY1, NR2A1, NR2A21, TCF, TCF14 Protein Size (# AA) 474 amino acids Molecular Weight 53kDa Product Format Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose. NCBI Gene Id 3172 Host Rabbit Clonality Polyclonal Concentration Batch dependent within range: 100 ul at 0.5 - 1 mg/ml Official Gene Full Hepatocyte nuclear factor 4, alpha Name Description This is a rabbit polyclonal antibody against HNF4A. It was validated on Western Blot by Aviva Systems Biology. At Aviva Systems Biology we manufacture rabbit polyclonal antibodies on a large scale (200-1000 products/month) of high throughput manner. Our antibodies are peptide based and protein family oriented. We usually provide antibodies covering each member of a whole protein family of your interest. We also use our best efforts to provide you antibodies recognize various epitopes of a target protein. For availability of antibody needed for your experiment, please inquire ([email protected]). Peptide Sequence Synthetic peptide located within the following region: MRLSKTLVDMDMADYSAALDPAYTTLEFENVQVLTMGNDTSPSEGTNLNA Description of The protein encoded by this gene is a nuclear transcription factor which binds DNA as a Target homodimer. The encoded protein controls the expression of several genes, including hepatocyte nuclear factor 1 alpha, a transcription factor which regulates the expression of several hepatic genes. -
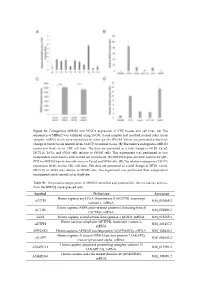
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared Amongst Δγ T, Innate Lymphoid, and Th Cells
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 δγ is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2016; 197:1460-1470; Prepublished online 6 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600643 http://www.jimmunol.org/content/197/4/1460 Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst T, Innate Lymphoid, and Th Cells You Jeong Lee, Gabriel J. Starrett, Seungeun Thera Lee, Rendong Yang, Christine M. Henzler, Stephen C. Jameson and Kristin A. Hogquist J Immunol cites 41 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2016/07/06/jimmunol.160064 3.DCSupplemental This article http://www.jimmunol.org/content/197/4/1460.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. The Journal of Immunology Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gd T, Innate Lymphoid, and Th Cells You Jeong Lee,* Gabriel J.