Addressing Incomplete Lineage Sorting and Paralogy in the Inference of Uncertain Salmonid Phylogenetic Relationships
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Eastslope Sculpin (Cottus Sp.) in Alberta
COSEWIC Assessment and Status Report on the "Eastslope" Sculpin Cottus sp. in Canada St. Mary and Milk River populations THREATENED 2005 COSEWIC COSEPAC COMMITTEE ON THE STATUS OF COMITÉ SUR LA SITUATION ENDANGERED WILDLIFE DES ESPÈCES EN PÉRIL IN CANADA AU CANADA COSEWIC status reports are working documents used in assigning the status of wildlife species suspected of being at risk. This report may be cited as follows: COSEWIC 2005. COSEWIC assessment and status report on the "eastslope" sculpin (St. Mary and Milk River population) Cottus sp. in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. vi + 30 pp. (www.sararegistry.gc.ca/status/status_e.cfm). Production note: This document is based on a report by Susan M. Pollard prepared for Alberta Sustainable Resource Development, Fish and Wildlife Division and the Alberta Conservation Association. The original report was published as Alberta Wildlife Status Report No. 51, February 2004, and is entitled Status of the St. Mary Shorthead Sculpin (provisionally Cottus bairdi punctulatus) in Alberta. Funding for the preparation of the original status report was provided by the Alberta Conservation Association and the Fish and Wildlife Division of Alberta Sustainable Resource Development. This document was overseen and edited by Bob Campbell, the COSEWIC Freshwater Fish Species Specialist Subcommittee Co- chair. For additional copies contact: COSEWIC Secretariat c/o Canadian Wildlife Service Environment Canada Ottawa, ON K1A 0H3 Tel.: (819) 997-4991 / (819) 953-3215 Fax: (819) 994-3684 E-mail: COSEWIC/[email protected] http://www.cosewic.gc.ca Ếgalement disponible en français sous le titre Ếvaluation et Rapport de situation du COSEPAC sur le chabot du versant est (populations des rivières St. -

The Phylogeny of Oncorhynchus (Euteleostei: Salmonidae) Based on Behavioral and Life History Characters
Copeia, 2007(3), pp. 520–533 The Phylogeny of Oncorhynchus (Euteleostei: Salmonidae) Based on Behavioral and Life History Characters MANU ESTEVE AND DEBORAH A. MCLENNAN There is no consensus between morphological and molecular data concerning the relationships within the Pacific basin salmon and trout clade Oncorhynchus. In this paper we add another source of characters to the discussion. Phylogenetic analysis of 39 behavioral and life history traits produced one tree structured (O. clarki (O. mykiss (O. masou (O. kisutch (O. tshawytscha (O. nerka (O. keta, O. gorbuscha))))))). This topology is congruent with the phylogeny based upon Bayesian analysis of all available nuclear and mitochondrial gene sequences, with the exception of two nodes: behavior supports the morphological data in breaking the sister-group relationship between O. mykiss and O. clarki, and between O. kisutch and O. tshawytscha. The behavioral traits agreed with molecular rather than morphological data in placing O. keta as the sister-group of O. gorbuscha. The behavioral traits also resolve the molecular-based ambiguity concerning the placement of O. masou, placing it as sister to the rest of the Pacific basin salmon. Behavioral plus morphological data placed Salmo, not Salvelinus, as more closely related to Oncorhynchus, but that placement was only weakly supported and awaits collection of missing data from enigmatic species such as the lake trout, Salvelinus namaycush. Overall, the phenotypic characters helped resolve ambiguities that may have been created by molecular introgression, while the molecular traits helped resolve ambiguities introduced by phenotypic homoplasy. It seems reasonable therefore, that systematists can best respond to the escalating biodiversity crisis by forming research groups to gather behavioral and ecological information while specimens are being collected for morphological and molecular analysis. -

Chromosomal Study of the Lenoks, Brachymystax(Salmoniformes
Journal of Species Research 2(1):91-98, 2013 Chromosomal study of the lenoks, Brachymystax (Salmoniformes, Salmonidae) from the South of the Russian Far East I.V. Kartavtseva*, L.K. Ginatulina, G.A. Nemkova and S.V. Shedko Institute of Biology and Soil Science of the Far East Branch of the Russian Academy of Sciences, Prospect 100 let Vladivostoku 159, Vladivostok 690022 *Correspondent: [email protected], [email protected] An investigation of the karyotypes of two species of the genus Brachymystax (B. lenok and B. tumensis) has been done for the Russia Primorye rivers running to the East Sea basin, and others belonging to Amur basin. Based on the analysis of two species chromosome characteristics, combined with original and literary data, four cytotypes have been described. One of these cytotypes (Cytotype I: 2n=90, NF=110-118) was the most common. This common cytotype belongs to B. tumensis from the rivers of the East Sea basin and B. lenok from the rivers of the Amur basin, i.e. extends to the zones of allopatry. In the rivers of the Amur river basin, in the zone of the sympatric habitat of two species, each taxon has karyotypes with different chromosome numbers, B. tumensis (2n=92) and B. lenok (2n=90). Because of the ability to determine a number of the chromosome arms for these two species, additional cytotype have been identified for B. tum- ensis: Cytotype II with 2n=92, NF=110-124 in the rivers basins of the Yellow sea and Amur river and for B. lenok three cytotypes: Cytotype I: 2n=90, NF=110 in the Amur river basin; Cytotype III with 2n=90, NF=106-126 in the Amur river basin and Cytotypes IV with 2n=92, NF=102 in the Baikal lake. -

Ecography ECOG-01937 Hattab, T., Leprieur, F., Ben Rais Lasram, F., Gravel, D., Le Loc’H, F
Ecography ECOG-01937 Hattab, T., Leprieur, F., Ben Rais Lasram, F., Gravel, D., Le Loc’h, F. and Albouy, C. 2016. Forecasting fine- scale changes in the food-web structure of coastal marine communities under climate change. – Ecography doi: 10.1111/ecog.01937 Supplementary material Forecasting fine-scale changes in the food-web structure of coastal marine communities under climate change by Hattab et al. Appendix 1 List of coastal exploited marine species considered in this study Species Genus Order Family Class Trophic guild Auxis rochei rochei (Risso, 1810) Auxis Perciformes Scombridae Actinopterygii Top predators Balistes capriscus Gmelin, 1789 Balistes Tetraodontiformes Balistidae Actinopterygii Macro-carnivorous Boops boops (Linnaeus, 1758) Boops Perciformes Sparidae Actinopterygii Basal species Carcharhinus plumbeus (Nardo, 1827) Carcharhinus Carcharhiniformes Carcharhinidae Elasmobranchii Top predators Dasyatis pastinaca (Linnaeus, 1758) Dasyatis Rajiformes Dasyatidae Elasmobranchii Top predators Dentex dentex (Linnaeus, 1758) Dentex Perciformes Sparidae Actinopterygii Macro-carnivorous Dentex maroccanus Valenciennes, 1830 Dentex Perciformes Sparidae Actinopterygii Macro-carnivorous Diplodus annularis (Linnaeus, 1758) Diplodus Perciformes Sparidae Actinopterygii Forage species Diplodus sargus sargus (Linnaeus, 1758) Diplodus Perciformes Sparidae Actinopterygii Macro-carnivorous (Geoffroy Saint- Diplodus vulgaris Hilaire, 1817) Diplodus Perciformes Sparidae Actinopterygii Basal species Engraulis encrasicolus (Linnaeus, 1758) Engraulis -
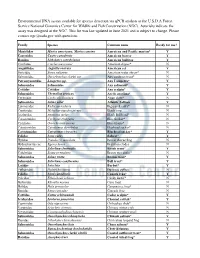
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

Acanthopterygii, Bone, Eurypterygii, Osteology, Percomprpha
Research in Zoology 2014, 4(2): 29-42 DOI: 10.5923/j.zoology.20140402.01 Comparative Osteology of the Jaws in Representatives of the Eurypterygian Fishes Yazdan Keivany Department of Natural Resources (Fisheries Division), Isfahan University of Technology, Isfahan, 84156-83111, Iran Abstract The osteology of the jaws in representatives of 49 genera in 40 families of eurypterygian fishes, including: Aulopiformes, Myctophiformes, Lampridiformes, Polymixiiformes, Percopsiformes, Mugiliformes, Atheriniformes, Beloniformes, Cyprinodontiformes, Stephanoberyciformes, Beryciformes, Zeiformes, Gasterosteiformes, Synbranchiformes, Scorpaeniformes (including Dactylopteridae), and Perciformes (including Elassomatidae) were studied. Generally, in this group, the upper jaw consists of the premaxilla, maxilla, and supramaxilla. The lower jaw consists of the dentary, anguloarticular, retroarticular, and sesamoid articular. In higher taxa, the premaxilla bears ascending, articular, and postmaxillary processes. The maxilla usually bears a ventral and a dorsal articular process. The supramaxilla is present only in some taxa. The dentary is usually toothed and bears coronoid and posteroventral processes. The retroarticular is small and located at the posteroventral corner of the anguloarticular. Keywords Acanthopterygii, Bone, Eurypterygii, Osteology, Percomprpha following method for clearing and staining bone and 1. Introduction cartilage provided in reference [18]. A camera lucida attached to a Wild M5 dissecting stereomicroscope was used Despite the introduction of modern techniques such as to prepare the drawings. The bones in the first figure of each DNA sequencing and barcoding, osteology, due to its anatomical section are arbitrarily shaded and labeled and in reliability, still plays an important role in the systematic the others are shaded in a consistent manner (dark, medium, study of fishes and comprises a major percent of today’s and clear) to facilitate comparison among the taxa. -

Revalidation and Redescription of Brachymystax Tsinlingensis Li, 1966 (Salmoniformes: Salmonidae) from China
Zootaxa 3962 (1): 191–205 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2015 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3962.1.12 http://zoobank.org/urn:lsid:zoobank.org:pub:F7864FFE-F182-455E-B37A-8A253D8DB72D Revalidation and redescription of Brachymystax tsinlingensis Li, 1966 (Salmoniformes: Salmonidae) from China YING-CHUN XING1,2, BIN-BIN LV3, EN-QI YE2, EN-YUAN FAN1, SHI-YANG LI4, LI-XIN WANG4, CHUN- GUANG ZHANG2,* & YA-HUI ZHAO2,* 1Natural Resource and Environment Research Center, Chinese Academy of Fishery Sciences, Beijing, China. 2Institute of Zoology, Chinese Academy of Sciences, Beijing, China. 3Yellow River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Xi’an, China. 4The College of Forestry of Beijing Forestry University, Beijing, China. *Corresponding authors: Yahui Zhao, [email protected]; Chunguang Zhang, [email protected] Abstract Brachymystax tsinlingensis Li, 1966 is revalidated and redescribed. It can be distinguished from all congeners by the fol- lowing combination of characteristics: no spots on operculum; gill rakers 15-20; lateral-line scales 98-116; pyloric caeca 60-71. Unique morphological characters and genetic divergence of this species are discussed. This species has a limited distribution in several streams of the middle part of the Qinling Mountains in China. Methods for management and pro- tection of B. tsinlingensis need to be re-evaluated. Key words: Brachymystax, revalidation, redescription, Salmonidae, China Introduction The genus Brachymystax Günther, 1866, belonging to Salmonidae, Salmoniformes, is distributed in eastern and northern Asia with three currently recognized valid species (Froese & Pauly, 2014): B. -

Morphology and Ultrastructure of Brachymystax Lenok Tsinlingensis
G Model YTICE-1010; No. of Pages 7 ARTICLE IN PRESS Tissue and Cell xxx (2016) xxx–xxx Contents lists available at ScienceDirect Tissue and Cell journal homepage: www.elsevier.com/locate/tice Morphology and ultrastructure of Brachymystax lenok tsinlingensis spermatozoa by scanning and transmission electron microscopy a,b,c c,d c,e c a,c,d,f,∗ Wei Guo , Jian Shao , Ping Li , Jinming Wu , Qiwei Wei a Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan 430072, China b University of Chinese Academy of Sciences, Beijing 100039, China c Key Laboratory of Freshwater Biodiversity Conservation, Ministry of Agriculture of China, Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences, No. 8, 1st Wudayuan Road, Donghu Hi-tech Development Zone, Wuhan 430223, China d College of Fisheries, Huazhong Agricultural University, Wuhan 430070, China e University of South Bohemia in Ceskéˇ Budejovice,ˇ Faculty of Fisheries and Protection of Waters, South Bohemian Research Center of Aquaculture and Biodiversity of Hydrocenoses, Research Institute of Fish Culture and Hydrobiology, Zátisíˇ 728/II, 389 25 Vodnany,ˇ Czech Republic f Freshwater Fisheries Research Center, Chinese Academy of Fisheries Science, Wuxi 214081, China a r t i c l e i n f o a b s t r a c t Article history: This study was conducted to investigate Brachymystax lenok tsinlingensis spermatozoa cell morphology Received 6 March 2016 and ultrastructure through scanning and transmission electron microscopy. Findings revealed that the Received in revised form 28 May 2016 spermatozoa can be differentiated into three major parts: a spherical head without an acrosome, a short Accepted 28 May 2016 mid-piece, and a long, cylindrical flagellum. -

Designating a Provincial Fossil
Designating a Provincial Fossil The Ministry of Forests, Lands, Natural Resource Operations and Rural Development is seeking input from the public on designating a Provincial Fossil to be added to the official Provincial Symbols of British Columbia. The designation of a Provincial fossil supports the principles of the Fossil Management Framework. The Framework recognizes fossils as important heritage resources with scientific and educational value. British Columbia’s list of official symbols and emblems include: Pacific Dogwood - adopted as the floral emblem in 1956 Jade - adopted as official gemstone in 1968 Stellar’s Jay - adopted as official bird in 1987 Western Red Cedar - adopted as official tree in 1988 Spirit Bear - adopted as the mammal emblem in 2006 Pacific Salmon - adopted as the fish emblem in 2013 Voting for your favourite fossil candidate Seven fossil candidates have been shortlisted through a public process in partnership with the British Columbia Paleontological Alliance (BCPA). The following criteria were used to select the fossil candidates: Be well known and easily recognizable; be more or less unique to British Columbia; reflect the unique geography of British Columbia; have wide appeal to a general audience; serve as an educational vehicle through which the biology, ecology, and geology of the time it represents can be made clear; and be amenable to designs for posters, displays and logos. The online voting process uses a web-based questionnaire tool, SurveyMonkey, allows one vote per computer profile and includes a simple verification step to avoid development of computer scripts for automatic votes. The Government of British Columbia will not collect, use or disclose personal information using SurveyMonkey. -

Imagine the Silver Beauty and the Fighting Spirit of Atlantic Salmon; The
Sakhalin Silver Text and Photos: Clemens Ratschan Imagine the silver beauty and the fighting spirit of Atlantic salmon; the complex, unpredictable life- history of sea trout and combine with the ferocious take and body mass of a predatory taimen. This will give you a glimpse of what fishing for Sakhalin taimen, the silver of the Russian Far East, is about. AM PLEASED TO introduce Siberian taimen, Hucho taimen. No this fish to the readers of wonder, scientists also erroneously Chasing Silver, because in related this far-eastern species to many respects it forms a the large-sized, non-anadromous missing link between the predators of the genus Hucho, which Ifishery for anadromous salmon and is a branch of the salmonoid tree for huchen, a big predatory non- that occurs exclusively in Eurasia. anadromous salmonoid in my home In Central Europe, Hucho hucho is country of Austria (‘Danube salmon’ restricted to the Danube System, in English. See article “Taimen” by where self-sustaining stocks are Wolfgang Hauer, issue 3/2010). presently only found in a handful of Sakhalin taimen is one of the rivers in Germany, Austria, Slovakia least-known salmonid species among and former Yugoslavia. Huchen is non-Russian fishermen; even many very closely related to the already- Russians tend to confuse it with the mentioned Siberian taimen. The latter | 62 | Chasing Silver Fly Fishing Magazine April’s Fav Five www.chasingsilvermagazine.com | 63 | Sakhalin Silver inhabits a distant, vast range from a habits. But one ecological feature expeditions to Japan. Later, the fish few places in European Russia to the is unique – all members of the true was assigned to the genus Parahucho, Lena and Amur rivers in the very far huchen live exclusively in fresh water, with regard to some obvious east of northern Asia. -

Mcabee Fossil Site Assessment
1 McAbee Fossil Site Assessment Final Report July 30, 2007 Revised August 5, 2007 Further revised October 24, 2008 Contract CCLAL08009 by Mark V. H. Wilson, Ph.D. Edmonton, Alberta, Canada Phone 780 435 6501; email [email protected] 2 Table of Contents Executive Summary ..............................................................................................................................................................3 McAbee Fossil Site Assessment ..........................................................................................................................................4 Introduction .......................................................................................................................................................................4 Geological Context ...........................................................................................................................................................8 Claim Use and Impact ....................................................................................................................................................10 Quality, Abundance, and Importance of the Fossils from McAbee ............................................................................11 Sale and Private Use of Fossils from McAbee..............................................................................................................12 Educational Use of Fossils from McAbee.....................................................................................................................13 -

XIV. Appendices
Appendix 1, Page 1 XIV. Appendices Appendix 1. Vertebrate Species of Alaska1 * Threatened/Endangered Fishes Scientific Name Common Name Eptatretus deani black hagfish Lampetra tridentata Pacific lamprey Lampetra camtschatica Arctic lamprey Lampetra alaskense Alaskan brook lamprey Lampetra ayresii river lamprey Lampetra richardsoni western brook lamprey Hydrolagus colliei spotted ratfish Prionace glauca blue shark Apristurus brunneus brown cat shark Lamna ditropis salmon shark Carcharodon carcharias white shark Cetorhinus maximus basking shark Hexanchus griseus bluntnose sixgill shark Somniosus pacificus Pacific sleeper shark Squalus acanthias spiny dogfish Raja binoculata big skate Raja rhina longnose skate Bathyraja parmifera Alaska skate Bathyraja aleutica Aleutian skate Bathyraja interrupta sandpaper skate Bathyraja lindbergi Commander skate Bathyraja abyssicola deepsea skate Bathyraja maculata whiteblotched skate Bathyraja minispinosa whitebrow skate Bathyraja trachura roughtail skate Bathyraja taranetzi mud skate Bathyraja violacea Okhotsk skate Acipenser medirostris green sturgeon Acipenser transmontanus white sturgeon Polyacanthonotus challengeri longnose tapirfish Synaphobranchus affinis slope cutthroat eel Histiobranchus bathybius deepwater cutthroat eel Avocettina infans blackline snipe eel Nemichthys scolopaceus slender snipe eel Alosa sapidissima American shad Clupea pallasii Pacific herring 1 This appendix lists the vertebrate species of Alaska, but it does not include subspecies, even though some of those are featured in the CWCS.