Genomics of Sablefish
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Eastslope Sculpin (Cottus Sp.) in Alberta
COSEWIC Assessment and Status Report on the "Eastslope" Sculpin Cottus sp. in Canada St. Mary and Milk River populations THREATENED 2005 COSEWIC COSEPAC COMMITTEE ON THE STATUS OF COMITÉ SUR LA SITUATION ENDANGERED WILDLIFE DES ESPÈCES EN PÉRIL IN CANADA AU CANADA COSEWIC status reports are working documents used in assigning the status of wildlife species suspected of being at risk. This report may be cited as follows: COSEWIC 2005. COSEWIC assessment and status report on the "eastslope" sculpin (St. Mary and Milk River population) Cottus sp. in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. vi + 30 pp. (www.sararegistry.gc.ca/status/status_e.cfm). Production note: This document is based on a report by Susan M. Pollard prepared for Alberta Sustainable Resource Development, Fish and Wildlife Division and the Alberta Conservation Association. The original report was published as Alberta Wildlife Status Report No. 51, February 2004, and is entitled Status of the St. Mary Shorthead Sculpin (provisionally Cottus bairdi punctulatus) in Alberta. Funding for the preparation of the original status report was provided by the Alberta Conservation Association and the Fish and Wildlife Division of Alberta Sustainable Resource Development. This document was overseen and edited by Bob Campbell, the COSEWIC Freshwater Fish Species Specialist Subcommittee Co- chair. For additional copies contact: COSEWIC Secretariat c/o Canadian Wildlife Service Environment Canada Ottawa, ON K1A 0H3 Tel.: (819) 997-4991 / (819) 953-3215 Fax: (819) 994-3684 E-mail: COSEWIC/[email protected] http://www.cosewic.gc.ca Ếgalement disponible en français sous le titre Ếvaluation et Rapport de situation du COSEPAC sur le chabot du versant est (populations des rivières St. -

Ecography ECOG-01937 Hattab, T., Leprieur, F., Ben Rais Lasram, F., Gravel, D., Le Loc’H, F
Ecography ECOG-01937 Hattab, T., Leprieur, F., Ben Rais Lasram, F., Gravel, D., Le Loc’h, F. and Albouy, C. 2016. Forecasting fine- scale changes in the food-web structure of coastal marine communities under climate change. – Ecography doi: 10.1111/ecog.01937 Supplementary material Forecasting fine-scale changes in the food-web structure of coastal marine communities under climate change by Hattab et al. Appendix 1 List of coastal exploited marine species considered in this study Species Genus Order Family Class Trophic guild Auxis rochei rochei (Risso, 1810) Auxis Perciformes Scombridae Actinopterygii Top predators Balistes capriscus Gmelin, 1789 Balistes Tetraodontiformes Balistidae Actinopterygii Macro-carnivorous Boops boops (Linnaeus, 1758) Boops Perciformes Sparidae Actinopterygii Basal species Carcharhinus plumbeus (Nardo, 1827) Carcharhinus Carcharhiniformes Carcharhinidae Elasmobranchii Top predators Dasyatis pastinaca (Linnaeus, 1758) Dasyatis Rajiformes Dasyatidae Elasmobranchii Top predators Dentex dentex (Linnaeus, 1758) Dentex Perciformes Sparidae Actinopterygii Macro-carnivorous Dentex maroccanus Valenciennes, 1830 Dentex Perciformes Sparidae Actinopterygii Macro-carnivorous Diplodus annularis (Linnaeus, 1758) Diplodus Perciformes Sparidae Actinopterygii Forage species Diplodus sargus sargus (Linnaeus, 1758) Diplodus Perciformes Sparidae Actinopterygii Macro-carnivorous (Geoffroy Saint- Diplodus vulgaris Hilaire, 1817) Diplodus Perciformes Sparidae Actinopterygii Basal species Engraulis encrasicolus (Linnaeus, 1758) Engraulis -
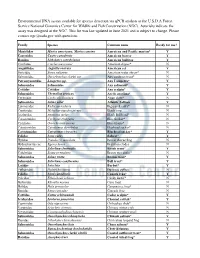
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

Acanthopterygii, Bone, Eurypterygii, Osteology, Percomprpha
Research in Zoology 2014, 4(2): 29-42 DOI: 10.5923/j.zoology.20140402.01 Comparative Osteology of the Jaws in Representatives of the Eurypterygian Fishes Yazdan Keivany Department of Natural Resources (Fisheries Division), Isfahan University of Technology, Isfahan, 84156-83111, Iran Abstract The osteology of the jaws in representatives of 49 genera in 40 families of eurypterygian fishes, including: Aulopiformes, Myctophiformes, Lampridiformes, Polymixiiformes, Percopsiformes, Mugiliformes, Atheriniformes, Beloniformes, Cyprinodontiformes, Stephanoberyciformes, Beryciformes, Zeiformes, Gasterosteiformes, Synbranchiformes, Scorpaeniformes (including Dactylopteridae), and Perciformes (including Elassomatidae) were studied. Generally, in this group, the upper jaw consists of the premaxilla, maxilla, and supramaxilla. The lower jaw consists of the dentary, anguloarticular, retroarticular, and sesamoid articular. In higher taxa, the premaxilla bears ascending, articular, and postmaxillary processes. The maxilla usually bears a ventral and a dorsal articular process. The supramaxilla is present only in some taxa. The dentary is usually toothed and bears coronoid and posteroventral processes. The retroarticular is small and located at the posteroventral corner of the anguloarticular. Keywords Acanthopterygii, Bone, Eurypterygii, Osteology, Percomprpha following method for clearing and staining bone and 1. Introduction cartilage provided in reference [18]. A camera lucida attached to a Wild M5 dissecting stereomicroscope was used Despite the introduction of modern techniques such as to prepare the drawings. The bones in the first figure of each DNA sequencing and barcoding, osteology, due to its anatomical section are arbitrarily shaded and labeled and in reliability, still plays an important role in the systematic the others are shaded in a consistent manner (dark, medium, study of fishes and comprises a major percent of today’s and clear) to facilitate comparison among the taxa. -

The Recent Distribution and Abundance of Non-Native Neogobius Fishes in the Slovak Section of the River Danube
J. Appl. Ichthyol. 21 (2005), 319–323 Received: January 30, 2005 Ó 2005 Blackwell Verlag, Berlin Accepted: June 15, 2005 ISSN 0175–8659 The recent distribution and abundance of non-native Neogobius fishes in the Slovak section of the River Danube By P. Jurajda1,J.Cˇ erny´2, M. Polacˇ ik1,3, Z. Valova´1,3, M. Jana´cˇ 1,3,R.Blazˇ ek3 and M. Ondracˇ kova´1 1Department of Fish Ecology, Institute of Vertebrate Biology AS CR, Brno, Czech Republic; 2Department of Hydrobiology, Institute of Zoology SAS, Bratislava, Slovak Republic; 3Department of Zoology and Ecology, Faculty of Science Masaryk University, Brno, Czech Republic Summary in the Sio´Channel (connecting Lake Balaton and the main The distributions of invasive Neogobius species were investi- Danube channel), and in the River Tisza and its tributary, gated in the Slovak section of the River Danube from the River Bodrog, both in Hungary (Ahnelt et al., 1998). Bratislava downstream to the village of Chl’aba. During More recently, monkey goby has been recorded in the October 2004, the main channel of the Danube was sampled, Slovakian Danube near Hungary (Stra´nˇ ai and Andreji, including by-pass, head-race and tail-race canals of the 2001) and a further upstream expansion of this species into Gabcˇ ı´kovo dam, backwaters and the lower-most sections of the Slovakian part of the River Bodrog can be expected the tributaries Maly´Dunaj, Hron, Va´h and IpelÕ. Three (Ahnelt et al., 1998; Kautman, 2001). Neogobius species already documented in Slovakia were An upstream expansion of racer goby has also been reported captured (monkey goby Neogobius fluviatilis, bighead goby in River Danube basin (summarized in Copp et al., 2005), N. -

Updated Checklist of Marine Fishes (Chordata: Craniata) from Portugal and the Proposed Extension of the Portuguese Continental Shelf
European Journal of Taxonomy 73: 1-73 ISSN 2118-9773 http://dx.doi.org/10.5852/ejt.2014.73 www.europeanjournaloftaxonomy.eu 2014 · Carneiro M. et al. This work is licensed under a Creative Commons Attribution 3.0 License. Monograph urn:lsid:zoobank.org:pub:9A5F217D-8E7B-448A-9CAB-2CCC9CC6F857 Updated checklist of marine fishes (Chordata: Craniata) from Portugal and the proposed extension of the Portuguese continental shelf Miguel CARNEIRO1,5, Rogélia MARTINS2,6, Monica LANDI*,3,7 & Filipe O. COSTA4,8 1,2 DIV-RP (Modelling and Management Fishery Resources Division), Instituto Português do Mar e da Atmosfera, Av. Brasilia 1449-006 Lisboa, Portugal. E-mail: [email protected], [email protected] 3,4 CBMA (Centre of Molecular and Environmental Biology), Department of Biology, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. E-mail: [email protected], [email protected] * corresponding author: [email protected] 5 urn:lsid:zoobank.org:author:90A98A50-327E-4648-9DCE-75709C7A2472 6 urn:lsid:zoobank.org:author:1EB6DE00-9E91-407C-B7C4-34F31F29FD88 7 urn:lsid:zoobank.org:author:6D3AC760-77F2-4CFA-B5C7-665CB07F4CEB 8 urn:lsid:zoobank.org:author:48E53CF3-71C8-403C-BECD-10B20B3C15B4 Abstract. The study of the Portuguese marine ichthyofauna has a long historical tradition, rooted back in the 18th Century. Here we present an annotated checklist of the marine fishes from Portuguese waters, including the area encompassed by the proposed extension of the Portuguese continental shelf and the Economic Exclusive Zone (EEZ). The list is based on historical literature records and taxon occurrence data obtained from natural history collections, together with new revisions and occurrences. -

Atlas of Marine Bony Fish Otoliths (Sagittae) of Southeastern-Southern
Original Article / Artigo Original Conversani et al.: Sagittae from the SouthwestBJOCE Atlantic Ocean Atlas of marine bony fish otoliths (sagittae) of Southeastern-Southern Brazil Part VII: Atheriniformes, Beloniformes, Beryciformes, Zeiformes, Syngnathiformes, Scorpaeniformes and Tetraodontiformes Valéria Regina Martins Conversani1, Marina Rito Brenha-Nunes1, César Santificetur1, Marcella Bockis Giaretta1, Carolina Correia Siliprandi1, Carmen Lucia Del Bianco Rossi-Wongtschowski1* 1 Instituto Oceanográfico da Universidade de São Paulo (Praça do Oceanográfico, 191, 05508-120 São Paulo, SP, Brazil) *Corresponding author: [email protected] ABSTRACT RESUMO In addition to the series of documents that we have Em adição à série de documentos que estamos been publishing on the "Atlas of Teleostei Otoliths publicando sobre o "Atlas de Otólitos para os peixes for the Southeastern-Southern Brazilian region", in Teleósteos da região Sudeste-Sul do Brasil", neste this volume we present the results of species of the volume apresentamos os resultados obtidos para espécies orders Atheriniformes (1 species), Beloniformes (5), das ordens Atheriniformes (1 espécie), Beloniformes Beryciformes (2), Zeiformes (2), Syngnathiformes (5), Beryciformes (2), Zeiformes (2), Syngnathiformes (2), Scorpaeniformes (9) and Tetraodontiformes (6). (2), Scorpaeniformes (9) e Tetraodontiformes (6). Foram Features, measurements and indices were analyzed analisadas as feições, medidas e índices usualmente according to methodology used in anterior series. empregados conforme metodologia -

Percomorph Phylogeny: a Survey of Acanthomorphs and a New Proposal
BULLETIN OF MARINE SCIENCE, 52(1): 554-626, 1993 PERCOMORPH PHYLOGENY: A SURVEY OF ACANTHOMORPHS AND A NEW PROPOSAL G. David Johnson and Colin Patterson ABSTRACT The interrelationships of acanthomorph fishes are reviewed. We recognize seven mono- phyletic terminal taxa among acanthomorphs: Lampridiformes, Polymixiiformes, Paracan- thopterygii, Stephanoberyciformes, Beryciformes, Zeiformes, and a new taxon named Smeg- mamorpha. The Percomorpha, as currently constituted, are polyphyletic, and the Perciformes are probably paraphyletic. The smegmamorphs comprise five subgroups: Synbranchiformes (Synbranchoidei and Mastacembeloidei), Mugilomorpha (Mugiloidei), Elassomatidae (Elas- soma), Gasterosteiformes, and Atherinomorpha. Monophyly of Lampridiformes is justified elsewhere; we have found no new characters to substantiate the monophyly of Polymixi- iformes (which is not in doubt) or Paracanthopterygii. Stephanoberyciformes uniquely share a modification of the extrascapular, and Beryciformes a modification of the anterior part of the supraorbital and infraorbital sensory canals, here named Jakubowski's organ. Our Zei- formes excludes the Caproidae, and characters are proposed to justify the monophyly of the group in that restricted sense. The Smegmamorpha are thought to be monophyletic principally because of the configuration of the first vertebra and its intermuscular bone. Within the Smegmamorpha, the Atherinomorpha and Mugilomorpha are shown to be monophyletic elsewhere. Our Gasterosteiformes includes the syngnathoids and the Pegasiformes -

Recycled Fish Sculpture (.PDF)
Recycled Fish Sculpture Name:__________ Fish: are a paraphyletic group of organisms that consist of all gill-bearing aquatic vertebrate animals that lack limbs with digits. At 32,000 species, fish exhibit greater species diversity than any other group of vertebrates. Sculpture: is three-dimensional artwork created by shaping or combining hard materials—typically stone such as marble—or metal, glass, or wood. Softer ("plastic") materials can also be used, such as clay, textiles, plastics, polymers and softer metals. They may be assembled such as by welding or gluing or by firing, molded or cast. Researched Photo Source: Alaskan Rainbow STEP ONE: CHOOSE one fish from the attached Fish Names list. Trout STEP TWO: RESEARCH on-line and complete the attached K/U Fish Research Sheet. STEP THREE: DRAW 3 conceptual sketches with colour pencil crayons of possible visual images that represent your researched fish. STEP FOUR: Once your fish designs are approved by the teacher, DRAW a representational outline of your fish on the 18 x24 and then add VALUE and COLOUR . CONSIDER: Individual shapes and forms for the various parts you will cut out of recycled pop aluminum cans (such as individual scales, gills, fins etc.) STEP FIVE: CUT OUT using scissors the various individual sections of your chosen fish from recycled pop aluminum cans. OVERLAY them on top of your 18 x 24 Representational Outline 18 x 24 Drawing representational drawing to judge the shape and size of each piece. STEP SIX: Once you have cut out all your shapes and forms, GLUE the various pieces together with a glue gun. -

A Checklist of the Fishes of the Monterey Bay Area Including Elkhorn Slough, the San Lorenzo, Pajaro and Salinas Rivers
f3/oC-4'( Contributions from the Moss Landing Marine Laboratories No. 26 Technical Publication 72-2 CASUC-MLML-TP-72-02 A CHECKLIST OF THE FISHES OF THE MONTEREY BAY AREA INCLUDING ELKHORN SLOUGH, THE SAN LORENZO, PAJARO AND SALINAS RIVERS by Gary E. Kukowski Sea Grant Research Assistant June 1972 LIBRARY Moss L8ndillg ,\:Jrine Laboratories r. O. Box 223 Moss Landing, Calif. 95039 This study was supported by National Sea Grant Program National Oceanic and Atmospheric Administration United States Department of Commerce - Grant No. 2-35137 to Moss Landing Marine Laboratories of the California State University at Fresno, Hayward, Sacramento, San Francisco, and San Jose Dr. Robert E. Arnal, Coordinator , ·./ "':., - 'I." ~:. 1"-"'00 ~~ ~~ IAbm>~toriesi Technical Publication 72-2: A GI-lliGKL.TST OF THE FISHES OF TtlE MONTEREY my Jl.REA INCLUDING mmORH SLOUGH, THE SAN LCRENZO, PAY-ARO AND SALINAS RIVERS .. 1&let~: Page 14 - A1estria§.·~iligtro1ophua - Stone cockscomb - r-m Page 17 - J:,iparis'W10pus." Ribbon' snailt'ish - HE , ,~ ~Ei 31 - AlectrlQ~iu.e,ctro1OphUfi- 87-B9 . .', . ': ". .' Page 31 - Ceb1diehtlrrs rlolaCewi - 89 , Page 35 - Liparis t!01:f-.e - 89 .Qhange: Page 11 - FmWulns parvipin¢.rl, add: Probable misidentification Page 20 - .BathopWuBt.lemin&, change to: .Mhgghilu§. llemipg+ Page 54 - Ji\mdJ11ui~~ add: Probable. misidentifioation Page 60 - Item. number 67, authOr should be .Hubbs, Clark TABLE OF CONTENTS INTRODUCTION 1 AREA OF COVERAGE 1 METHODS OF LITERATURE SEARCH 2 EXPLANATION OF CHECKLIST 2 ACKNOWLEDGEMENTS 4 TABLE 1 -

Order SCORPAENIFORMES DACTYLOPTERIDAE Dactylopterus Volitans (Linnaeus, 1758)
click for previous page 1230 Bony Fishes Order SCORPAENIFORMES DACTYLOPTERIDAE Flying gurnards by W.F. Smith-Vaniz, US Geological Survey, Florida, USA A single species occurring in the area. Dactylopterus volitans (Linnaeus, 1758) Frequent synonyms / misidentifications: None / None. FAO names: En - Flying gurnard; Fr - Poule de mer; Sp - Alón. iagnostic characters: A moderately elongate fish. Head blunt, top and sides encased in a bony Dshield; a long, keeled spine extending posteriorly from the nape to below midbase of first dorsal fin; angle of preopercle also bearing a long spine, with a serrate keel; jaws with a band of small nodular teeth. Spinous and soft dorsal fins separated by a deep notch; anterior 2 dorsal-fin spines adjacent to each other, interconnected by a basal membrane, and not separated from remainder of spinous dorsal fin; anal fin with only 6 soft rays; caudal fin emarginate, with 2 sharp keels on its base; bases of pectoral fins hori- zontal, the fins divided into 2 sections, an anterior short part of 6 soft rays and a posterior long part of 26 to 30 soft rays which reach the caudal-fin base in adults. Scales scute-like with sharp keels. Colour: variable with surroundings; often yellowish brown, with bright blue spots on pectoral fins. Similar species occurring in the area The bony head shield in combination with the long keeled spine on nape and the long serrated preopercular spine will readily distinguish this species from all others occurring in the area. The Triglidae, which are superfi- cially similar in having large pectoral fins and the head encased in bony armour, differ additionally in having the spinous dorsal fin entire and all spines united by a fin membrane. -
Columbia Sculpin (Cottus Hubbsi) Is a Small, Freshwater Sculpin (Cottidae)
COSEWIC Assessment and Status Report on the Columbia Sculpin Cottus hubbsi in Canada SPECIAL CONCERN 2010 COSEWIC status reports are working documents used in assigning the status of wildlife species suspected of being at risk. This report may be cited as follows: COSEWIC. 2010. COSEWIC assessment and status report on the Columbia Sculpin Cottus hubbsi in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. xii + 32 pp. (www.sararegistry.gc.ca/status/status_e.cfm). Production note: COSEWIC acknowledges Don McPhail for writing the provisional status report on the Columbia Sculpin, Cottus hubbsi, prepared under contract with Environment Canada. The contractor’s involvement with the writing of the status report ended with the acceptance of the provisional report. Any modifications to the status report during the subsequent preparation of the 6-month interim status report and 2-month interim status reports were overseen by Dr. Eric Taylor, COSEWIC Freshwater Fishes Specialist Subcommittee Co-chair. For additional copies contact: COSEWIC Secretariat c/o Canadian Wildlife Service Environment Canada Ottawa, ON K1A 0H3 Tel.: 819-953-3215 Fax: 819-994-3684 E-mail: COSEWIC/[email protected] http://www.cosewic.gc.ca Également disponible en français sous le titre Ếvaluation et Rapport de situation du COSEPAC sur le chabot du Columbia (Cottus hubbsi) au Canada. Cover illustration/photo: Columbia Sculpin — illustration by Diana McPhail. Her Majesty the Queen in Right of Canada, 2011. Catalogue No. CW69-14/268-2011E-PDF ISBN 978-1-100-18590-3 Recycled paper COSEWIC Assessment Summary Assessment Summary – November 2010 Common name Columbia Sculpin Scientific name Cottus hubbsi Status Special Concern Reason for designation In Canada, this small freshwater fish is endemic to the Columbia River basin where it has a small geographic distribution.