Association of Variably Methylated Tumour DNA Regions with Overall
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Histone-Related Genes Are Hypermethylated in Lung Cancer
Published OnlineFirst October 1, 2019; DOI: 10.1158/0008-5472.CAN-19-1019 Cancer Genome and Epigenome Research Histone-Related Genes Are Hypermethylated in Lung Cancer and Hypermethylated HIST1H4F Could Serve as a Pan-Cancer Biomarker Shihua Dong1,Wei Li1, Lin Wang2, Jie Hu3,Yuanlin Song3, Baolong Zhang1, Xiaoguang Ren1, Shimeng Ji3, Jin Li1, Peng Xu1, Ying Liang1, Gang Chen4, Jia-Tao Lou2, and Wenqiang Yu1 Abstract Lung cancer is the leading cause of cancer-related deaths lated in all 17 tumor types from TCGA datasets (n ¼ 7,344), worldwide. Cytologic examination is the current "gold stan- which was further validated in nine different types of cancer dard" for lung cancer diagnosis, however, this has low sensi- (n ¼ 243). These results demonstrate that HIST1H4F can tivity. Here, we identified a typical methylation signature of function as a universal-cancer-only methylation (UCOM) histone genes in lung cancer by whole-genome DNA methyl- marker, which may aid in understanding general tumorigen- ation analysis, which was validated by The Cancer Genome esis and improve screening for early cancer diagnosis. Atlas (TCGA) lung cancer cohort (n ¼ 907) and was further confirmed in 265 bronchoalveolar lavage fluid samples with Significance: These findings identify a new biomarker for specificity and sensitivity of 96.7% and 87.0%, respectively. cancer detection and show that hypermethylation of histone- More importantly, HIST1H4F was universally hypermethy- related genes seems to persist across cancers. Introduction to its low specificity, LDCT is far from satisfactory as a screening tool for clinical application, similar to other currently used cancer Lung cancer is one of the most common malignant tumors and biomarkers, such as carcinoembryonic antigen (CEA), neuron- the leading cause of cancer-related deaths worldwide (1, 2). -

Genome-Wide DNA Methylation Analysis Reveals Molecular Subtypes of Pancreatic Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 17), pp: 28990-29012 Research Paper Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer Nitish Kumar Mishra1 and Chittibabu Guda1,2,3,4 1Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE, 68198, USA 2Bioinformatics and Systems Biology Core, University of Nebraska Medical Center, Omaha, NE, 68198, USA 3Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE, 68198, USA 4Fred and Pamela Buffet Cancer Center, University of Nebraska Medical Center, Omaha, NE, 68198, USA Correspondence to: Chittibabu Guda, email: [email protected] Keywords: TCGA, pancreatic cancer, differential methylation, integrative analysis, molecular subtypes Received: October 20, 2016 Accepted: February 12, 2017 Published: March 07, 2017 Copyright: Mishra et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Pancreatic cancer (PC) is the fourth leading cause of cancer deaths in the United States with a five-year patient survival rate of only 6%. Early detection and treatment of this disease is hampered due to lack of reliable diagnostic and prognostic markers. Recent studies have shown that dynamic changes in the global DNA methylation and gene expression patterns play key roles in the PC development; hence, provide valuable insights for better understanding the initiation and progression of PC. In the current study, we used DNA methylation, gene expression, copy number, mutational and clinical data from pancreatic patients. -
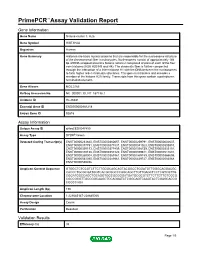
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 3, H2a Gene Symbol HIST3H2A Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A H2B H3 and H4). The chromatin fiber is further compacted through the interaction of a linker histone H1 with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene contain a palindromic termination element. Gene Aliases MGC3165 RefSeq Accession No. NC_000001.10, NT_167186.1 UniGene ID Hs.26331 Ensembl Gene ID ENSG00000181218 Entrez Gene ID 92815 Assay Information Unique Assay ID qHsaCED0047450 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233840, ENST00000360657, ENST00000259791, ENST00000602637, ENST00000377791, ENST00000377831, ENST00000341023, ENST00000303910, ENST00000359193, ENST00000377459, ENST00000358739, ENST00000333151, ENST00000330180, ENST00000357320, ENST00000359611, ENST00000511601, ENST00000260008, ENST00000369161, ENST00000369159, ENST00000366695, ENST00000543095, ENST00000389961, ENST00000439727, ENST00000583968, ENST00000580456 Amplicon Context Sequence GTGGCTCTCCGTCTTCTTGGGCAGCAGTACGGCCTGGATGTTGGGCAGGACGC CACCCTGCGCGATGGTCACGCGGCCCAGCAGCTTGTTGAGCTCCTCGTCGTTG CGGATGGCCAGCTGCAGGTGGCGCGGGATGATGCGCGTCTTCTTGTTGTCGCG -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap18559c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP18559c Specification HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) - Product Information Application WB,E Primary Accession Q93079 Other Accession P57053, Q9PSW9, P0C1H5, P0C1H4, Q6PC60, Q8CGP0, Q8N257, Q9D2U9, Q5QNW6, Q64524, Q16778, Q64525, Q00715, Q5BJA5, P0C1H3, P62808, Q8CGP2, P23527, Q99877, Q32L48, P10854, Q99879, Q99880, Q8CGP1, O60814, Q2M2T1, HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody P06899, Q64478, (Center) (Cat. #AP18559c) western blot P10853, P58876, analysis in MCF-7 cell line lysates Q6ZWY9, P62807 (35ug/lane).This demonstrates the Reactivity Human HIST1H2BH/HIST1H2BK/HIST3H2BB antibody Predicted Xenopus, Mouse, detected the Bovine, Chicken, HIST1H2BH/HIST1H2BK/HIST3H2BB protein Zebrafish, Rat (arrow). Host Rabbit Clonality Polyclonal Isotype Rabbit Ig HIST1H2BH/HIST1H2BK/HIST3H2BB Calculated MW 13892 Antibody (Center) - Background Antigen Region 26-52 Histones are basic nuclear proteins that are HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody responsible (Center) - Additional Information for the nucleosome structure of the chromosomal fiber in Gene ID 8345 eukaryotes. Two molecules of each of the four core histones (H2A, Other Names H2B, H3, and H4) form an octamer, around Histone H2B type 1-H, Histone H2Bj, H2B/j, which approximately 146 bp HIST1H2BH, H2BFJ of DNA is wrapped in repeating units, called nucleosomes. The Target/Specificity linker histone, H1, interacts with linker DNA This HIST1H2BH/HIST1H2BK/HIST3H2BB between nucleosomes antibody is generated from rabbits and functions in the compaction of chromatin immunized with a KLH conjugated synthetic into higher order peptide between 26-52 amino acids from structures. This gene is intronless and encodes the Central region of human a member of the HIST1H2BH/HIST1H2BK/HIST3H2BB. -

A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder
INVESTIGATION A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder Aparna Prasad,* Daniele Merico,* Bhooma Thiruvahindrapuram,* John Wei,* Anath C. Lionel,*,† Daisuke Sato,* Jessica Rickaby,* Chao Lu,* Peter Szatmari,‡ Wendy Roberts,§ Bridget A. Fernandez,** Christian R. Marshall,*,†† Eli Hatchwell,‡‡ Peggy S. Eis,‡‡ and Stephen W. Scherer*,†,††,1 *The Centre for Applied Genomics, Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto M5G 1L7, Canada, †Department of Molecular Genetics, University of Toronto, Toronto M5G 1L7, Canada, ‡Offord Centre for Child Studies, Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton L8P 3B6, § Canada, Autism Research Unit, The Hospital for Sick Children, Toronto M5G 1X8, Canada, **Disciplines of Genetics and Medicine, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3V6, Canada, ††McLaughlin Centre, University of Toronto, Toronto M5G 1L7, Canada, and ‡‡Population Diagnostics, Inc., Melville, New York 11747 ABSTRACT The identification of rare inherited and de novo copy number variations (CNVs) in human KEYWORDS subjects has proven a productive approach to highlight risk genes for autism spectrum disorder (ASD). A rare variants variety of microarrays are available to detect CNVs, including single-nucleotide polymorphism (SNP) arrays gene copy and comparative genomic hybridization (CGH) arrays. Here, we examine a cohort of 696 unrelated ASD number cases using a high-resolution one-million feature CGH microarray, the majority of which were previously chromosomal genotyped with SNP arrays. Our objective was to discover new CNVs in ASD cases that were not detected abnormalities by SNP microarray analysis and to delineate novel ASD risk loci via combined analysis of CGH and SNP array cytogenetics data sets on the ASD cohort and CGH data on an additional 1000 control samples. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Chew Et Al-2021-Nature Communi
Short H2A histone variants are expressed in cancer Guo-Liang Chew, Marie Bleakley, Robert Bradley, Harmit Malik, Steven Henikoff, Antoine Molaro, Jay Sarthy To cite this version: Guo-Liang Chew, Marie Bleakley, Robert Bradley, Harmit Malik, Steven Henikoff, et al.. Short H2A histone variants are expressed in cancer. Nature Communications, Nature Publishing Group, 2021, 12 (1), pp.490. 10.1038/s41467-020-20707-x. hal-03118929 HAL Id: hal-03118929 https://hal.archives-ouvertes.fr/hal-03118929 Submitted on 22 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ARTICLE https://doi.org/10.1038/s41467-020-20707-x OPEN Short H2A histone variants are expressed in cancer Guo-Liang Chew 1, Marie Bleakley2, Robert K. Bradley 3,4,5, Harmit S. Malik4,6, Steven Henikoff 4,6, ✉ ✉ Antoine Molaro 4,7 & Jay Sarthy 4 Short H2A (sH2A) histone variants are primarily expressed in the testes of placental mammals. Their incorporation into chromatin is associated with nucleosome destabilization and modulation of alternate splicing. Here, we show that sH2As innately possess features similar to recurrent oncohistone mutations associated with nucleosome instability. Through 1234567890():,; analyses of existing cancer genomics datasets, we find aberrant sH2A upregulation in a broad array of cancers, which manifest splicing patterns consistent with global nucleosome destabilization. -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 RESEARCH ARTICLE A Mutation in Histone H2B Represents a New Class of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,4, Rajarshi P. Ghosh5, Adrian T. Grzybowski6, Tao Yu7, Qiao Zhang8, Alberto Riva9, Tanmay P. Lele8, George C. Schatz4, Neil L. Kelleher4, Alexander J. Ruthenburg6, Jan Liphardt5, and Jonathan D. Licht1 Downloaded from cancerdiscovery.aacrjournals.org on September 30, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 ABSTRACT By examination of the cancer genomics database, we identified a new set of mutations in core histones that frequently recur in cancer patient samples and are predicted to disrupt nucleosome stability. In support of this idea, we characterized a glutamate to lysine mutation of histone H2B at amino acid 76 (H2B-E76K), found particularly in bladder and head and neck cancers, that disrupts the interaction between H2B and H4. Although H2B-E76K forms dimers with H2A, it does not form stable histone octamers with H3 and H4 in vitro, and when recon- stituted with DNA forms unstable nucleosomes with increased sensitivity to nuclease. Expression of the equivalent H2B mutant in yeast restricted growth at high temperature and led to defective nucleosome-mediated gene repression. Significantly, H2B-E76K expression in the normal mammary epithelial cell line MCF10A increased cellular proliferation, cooperated with mutant PIK3CA to pro- mote colony formation, and caused a significant drift in gene expression and fundamental changes in chromatin accessibility, particularly at gene regulatory elements. -

1 Global Analysis of Gene Expression Reveals Mrna Superinduction Is
1 2 3 4 5 Global analysis of gene expression reveals mRNA superinduction is required for the 6 inducible immune response to a bacterial pathogen 7 8 9 10 Kevin C. Barry1, Nicholas T. Ingolia2*, and Russell E. Vance1,3,4* 11 12 13 14 1Division of Immunology and Pathogenesis, Department of Molecular & Cell Biology 15 2Division of Biochemistry, Biophysics and Structural Biology, Department of Molecular 16 & Cell Biology 17 3Cancer Research Laboratory 18 4Howard Hughes Medical Institute 19 University of California, Berkeley, California 94720, USA. 20 21 22 23 *Correspondence: Russell E. Vance, Division of Immunology and Pathogenesis, 24 Department of Molecular and Cell Biology, University of California, Berkeley, CA 25 94720-3200. Phone: (510) 643-2795. Fax: (510) 642-1386. E-mail: [email protected] 26 or Nicholas T. Ingolia, Division of Biochemistry, Biophysics and Structural Biology 27 Department of Molecular and Cell Biology, University of California, Berkeley, CA 28 94720-3200. Phone: (510) 664-7071. Email: [email protected] 29 1 30 Abstract 31 The inducible innate immune response to infection requires a concerted process of gene 32 expression that is regulated at multiple levels. Most global analyses of the innate immune 33 response have focused on transcription induced by defined immunostimulatory ligands, 34 such as lipopolysaccharide. However, the response to pathogens involves additional 35 complexity, as pathogens interfere with virtually every step of gene expression. How 36 cells respond to pathogen-mediated disruption of gene expression to nevertheless initiate 37 protective responses remains unclear. We previously discovered that a pathogen- 38 mediated blockade of host protein synthesis provokes the production of specific pro- 39 inflammatory cytokines. -

Noncoding Rnas As Novel Pancreatic Cancer Targets
NONCODING RNAS AS NOVEL PANCREATIC CANCER TARGETS by Amy Makler A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, FL August 2018 Copyright 2018 by Amy Makler ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous support, constant encouragement, and his gentle, but sometimes critical, guidance throughout the past two years of my master’s education. His faith in my abilities and his belief in my future success ensured I continue down this path of research. Working in Dr. Narayanan’s lab has truly been an unforgettable experience as well as a critical step in my future endeavors. I would also like to extend my gratitude to my committee members, Dr. Binninger and Dr. Jia, for their support and suggestions regarding my thesis. Their recommendations added a fresh perspective that enriched our initial hypothesis. They have been indispensable as members of my committee, and I thank them for their contributions. My parents have been integral to my successes in life and their support throughout my education has been crucial. They taught me to push through difficulties and encouraged me to pursue my interests. Thank you, mom and dad! I would like to thank my boyfriend, Joshua Disatham, for his assistance in ensuring my writing maintained a logical progression and flow as well as his unwavering support. He was my rock when the stress grew unbearable and his encouraging words kept me pushing along. -

The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,†
bioRxiv preprint doi: https://doi.org/10.1101/720235; this version posted July 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,† Abstract Core histone proteins H2A, H2B, H3, and H4 are encoded by a large family of genes dis- tributed across the human genome. Canonical core histones contribute the majority of proteins to bulk chromatin packaging, and are encoded in 4 clusters by 65 coding genes comprising 17 for H2A, 18 for H2B, 15 for H3, and 15 for H4, along with at least 17 total pseudogenes. The canonical core histone genes display coding variation that gives rise to 11 H2A, 15 H2B, 4 H3, and 2 H4 unique protein isoforms. Although histone proteins are highly conserved overall, these isoforms represent a surprising and seldom recognised variation with amino acid identity as low as 77 % between canonical histone proteins of the same type. The gene sequence and protein isoform diversity also exceeds com- monly used subtype designations such as H2A.1 and H3.1, and exists in parallel with the well-known specialisation of variant histone proteins. RNA sequencing of histone transcripts shows evidence for differential expression of histone genes but the functional significance of this variation has not yet been investigated. To assist understanding of the implications of histone gene and protein diversity we have catalogued the entire human canonical core histone gene and protein complement.