1471-2164-6-108.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Focus on the Small Heat Shock Protein HSPB1 Autofagie in De Erfelij
Faculteit Faculteit Farmaceutische, Biomedische en Diergeneeskundige wetenschappen Biochemie en Biotechnologie Autophagy in inherited peripheral neuropathies: Focus on the small heat shock protein HSPB1 Autofagie in de erfelijke perifere neuropathieën: Focus op de kleine heat shock proteïne HSPB1 Proefschrift voorgelegd tot het behalen van de graad van Doctor in de Wetenschappen: Biochemie en Biotechnologie aan de Universiteit Antwerpen. te verdedigen door Mansour HAIDAR Promotor Prof. Dr. Vincent Timmerman Antwerpen, 2018 1 2 “Haud igitur redit ad Nihilum res ulla, sed omnes Discidio redeunt in corpora materiai” Lucretius, De Rerum Natura, Book I. 250 3 4 Members of the jury Chair Prof. Dr. Wim Vanden Berghe, PhD (UA, Antwerp, Belgium) Promotor Prof. Dr. Vincent Timmerman, PhD (UA, Antwerp, Belgium) Internal jury member Prof. Dr. Wim Martinet, PhD (UA, Antwerp, Belgium) External jury members Prof. Dr. Joy Irobi (UHasselt, Hasselt, Belgium) Prof. Dr. Maurizio D’Antonio (San Raffaele Institute, Milan, Italy) Prof. Dr. Ir. Winnok De Vos (UA, Antwerp, Belgium) 5 6 Table of Contents Summary/Samenvatting 9 Rationale and Aims 13 Introduction Chapter 1 Autophagy as an emerging common pathomechanism in inherited 15 peripheral neuropathies Chapter 2 Small heat shock proteins: Their role in proteostasis 79 and neurodegeneration Results Chapter 3 HSPB1 is required for Autophagy: Insights from CMT-causing mutations 103 Chapter 4 An interactomics study of HSPB1 wild-type and mutant links it to the 129 autophagy receptor P62 Discussion 179 List of abbreviations 195 Curriculum Vitae 199 Acknowledgements 203 7 8 Summary Inherited peripheral neuropathies (IPNs) are genetically heterogeneous disorders affecting mainly the peripheral nervous system and with over 1500 mutations in more than 80 affected genes discovered so far. -

Supporting Information
Supporting Information Edgar et al. 10.1073/pnas.1601895113 SI Methods (Actimetrics), and recordings were analyzed using LumiCycle Mice. Sample size was determined using the resource equation: Data Analysis software (Actimetrics). E (degrees of freedom in ANOVA) = (total number of exper- – Cell Cycle Analysis of Confluent Cell Monolayers. NIH 3T3, primary imental animals) (number of experimental groups), with −/− sample size adhering to the condition 10 < E < 20. For com- WT, and Bmal1 fibroblasts were sequentially transduced − − parison of MuHV-4 and HSV-1 infection in WT vs. Bmal1 / with lentiviral fluorescent ubiquitin-based cell cycle indicators mice at ZT7 (Fig. 2), the investigator did not know the genotype (FUCCI) mCherry::Cdt1 and amCyan::Geminin reporters (32). of the animals when conducting infections, bioluminescence Dual reporter-positive cells were selected by FACS (Influx Cell imaging, and quantification. For bioluminescence imaging, Sorter; BD Biosciences) and seeded onto 35-mm dishes for mice were injected intraperitoneally with endotoxin-free lucif- subsequent analysis. To confirm that expression of mCherry:: Cdt1 and amCyan::Geminin correspond to G1 (2n DNA con- erin (Promega E6552) using 2 mg total per mouse. Following < ≤ anesthesia with isofluorane, they were scanned with an IVIS tent) and S/G2 (2 n 4 DNA content) cell cycle phases, Lumina (Caliper Life Sciences), 15 min after luciferin admin- respectively, cells were stained with DNA dye DRAQ5 (abcam) and analyzed by flow cytometry (LSR-Fortessa; BD Biosci- istration. Signal intensity was quantified using Living Image ences). To examine dynamics of replicative activity under ex- software (Caliper Life Sciences), obtaining maximum radiance perimental confluent conditions, synchronized FUCCI reporter for designated regions of interest (photons per second per − − − monolayers were observed by time-lapse live cell imaging over square centimeter per Steradian: photons·s 1·cm 2·sr 1), relative 3 d (Nikon Eclipse Ti-E inverted epifluorescent microscope). -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Multi-Sample Full-Length Transcriptome Analysis of 22 Breast Cancer Clinical
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Multi-sample Full-length Transcriptome Analysis of 22 Breast Cancer Clinical Specimens with Long-Read Sequencing Shinichi Namba1, §, Toshihide Ueno1, Shinya Kojima1, Yosuke Tanaka1, Satoshi Inoue1, Fumishi Kishigami1, Noriko Maeda2, Tomoko Ogawa3, Shoichi Hazama4, Yuichi Shiraishi5, Hiroyuki Mano1, and Masahito Kawazu1* Author affiliations: 1Division of Cellular Signaling, 5Division of Genome Analysis Platform Development, Research Institute, National Cancer Center, Tokyo 104-0045, Japan; 2Department of Gastroenterological, Breast and Endocrine Surgery, 4Department of Translational Research and Developmental Therapeutics against Cancer, Yamaguchi University Graduate School of Medicine, Yamaguchi 755-8505, Japan; 3Department of Breast Surgery, Mie University Hospital, Mie 514-8507, Japan; § Present address, Department of Statistical Genetics, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan; * Corresponding author E-mail:[email protected] (MK) bioRxiv preprint doi: https://doi.org/10.1101/2020.07.15.199851; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Although transcriptome alteration is considered as one of the essential drivers of carcinogenesis, conventional short-read RNAseq technology has limited researchers from directly exploring full-length transcripts, only focusing on individual splice sites. -
![A Genomic Atlas of Human Adrenal and Gonad Development [Version 2; Referees: 4 Approved] Ignacio Del Valle1, Federica Buonocore1, Andrew J](https://docslib.b-cdn.net/cover/1314/a-genomic-atlas-of-human-adrenal-and-gonad-development-version-2-referees-4-approved-ignacio-del-valle1-federica-buonocore1-andrew-j-711314.webp)
A Genomic Atlas of Human Adrenal and Gonad Development [Version 2; Referees: 4 Approved] Ignacio Del Valle1, Federica Buonocore1, Andrew J
Wellcome Open Research 2017, 2:25 Last updated: 08 NOV 2017 RESEARCH ARTICLE A genomic atlas of human adrenal and gonad development [version 2; referees: 4 approved] Ignacio del Valle1, Federica Buonocore1, Andrew J. Duncan1, Lin Lin1, Martino Barenco2, Rahul Parnaik1, Sonia Shah3,4, Mike Hubank5, Dianne Gerrelli2, John C. Achermann 1 1Genetics and Genomic Medicine, UCL Great Ormond Street Institute of Child Health, London, UK 2Developmental Biology and Cancer, UCL Great Ormond Street Institute of Child Health, London, UK 3Institute for Molecular Bioscience, University of Queensland, Brisbane, Australia 4Institute of Cardiovascular Science, University College London, London, UK 5The Centre for Molecular Pathology, Royal Marsden Hospital, Sutton, UK v2 First published: 07 Apr 2017, 2:25 (doi: 10.12688/wellcomeopenres.11253.1) Open Peer Review Latest published: 23 Oct 2017, 2:25 (doi: 10.12688/wellcomeopenres.11253.2) Referee Status: Abstract Background: In humans, the adrenal glands and gonads undergo distinct biological events between 6-10 weeks post conception (wpc), such as testis Invited Referees determination, the onset of steroidogenesis and primordial germ cell 1 2 3 4 development. However, relatively little is currently known about the genetic mechanisms underlying these processes. We therefore aimed to generate a detailed genomic atlas of adrenal and gonad development across these critical version 2 report report stages of human embryonic and fetal development. published Methods: RNA was extracted from 53 tissue samples between 6-10 wpc 23 Oct 2017 (adrenal, testis, ovary and control). Affymetrix array analysis was performed and differential gene expression was analysed using Bioconductor. A version 1 mathematical model was constructed to investigate time-series changes across published report report report report 07 Apr 2017 the dataset. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -
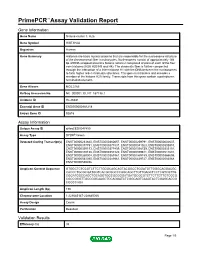
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 3, H2a Gene Symbol HIST3H2A Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A H2B H3 and H4). The chromatin fiber is further compacted through the interaction of a linker histone H1 with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene contain a palindromic termination element. Gene Aliases MGC3165 RefSeq Accession No. NC_000001.10, NT_167186.1 UniGene ID Hs.26331 Ensembl Gene ID ENSG00000181218 Entrez Gene ID 92815 Assay Information Unique Assay ID qHsaCED0047450 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233840, ENST00000360657, ENST00000259791, ENST00000602637, ENST00000377791, ENST00000377831, ENST00000341023, ENST00000303910, ENST00000359193, ENST00000377459, ENST00000358739, ENST00000333151, ENST00000330180, ENST00000357320, ENST00000359611, ENST00000511601, ENST00000260008, ENST00000369161, ENST00000369159, ENST00000366695, ENST00000543095, ENST00000389961, ENST00000439727, ENST00000583968, ENST00000580456 Amplicon Context Sequence GTGGCTCTCCGTCTTCTTGGGCAGCAGTACGGCCTGGATGTTGGGCAGGACGC CACCCTGCGCGATGGTCACGCGGCCCAGCAGCTTGTTGAGCTCCTCGTCGTTG CGGATGGCCAGCTGCAGGTGGCGCGGGATGATGCGCGTCTTCTTGTTGTCGCG -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder
INVESTIGATION A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder Aparna Prasad,* Daniele Merico,* Bhooma Thiruvahindrapuram,* John Wei,* Anath C. Lionel,*,† Daisuke Sato,* Jessica Rickaby,* Chao Lu,* Peter Szatmari,‡ Wendy Roberts,§ Bridget A. Fernandez,** Christian R. Marshall,*,†† Eli Hatchwell,‡‡ Peggy S. Eis,‡‡ and Stephen W. Scherer*,†,††,1 *The Centre for Applied Genomics, Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto M5G 1L7, Canada, †Department of Molecular Genetics, University of Toronto, Toronto M5G 1L7, Canada, ‡Offord Centre for Child Studies, Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton L8P 3B6, § Canada, Autism Research Unit, The Hospital for Sick Children, Toronto M5G 1X8, Canada, **Disciplines of Genetics and Medicine, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3V6, Canada, ††McLaughlin Centre, University of Toronto, Toronto M5G 1L7, Canada, and ‡‡Population Diagnostics, Inc., Melville, New York 11747 ABSTRACT The identification of rare inherited and de novo copy number variations (CNVs) in human KEYWORDS subjects has proven a productive approach to highlight risk genes for autism spectrum disorder (ASD). A rare variants variety of microarrays are available to detect CNVs, including single-nucleotide polymorphism (SNP) arrays gene copy and comparative genomic hybridization (CGH) arrays. Here, we examine a cohort of 696 unrelated ASD number cases using a high-resolution one-million feature CGH microarray, the majority of which were previously chromosomal genotyped with SNP arrays. Our objective was to discover new CNVs in ASD cases that were not detected abnormalities by SNP microarray analysis and to delineate novel ASD risk loci via combined analysis of CGH and SNP array cytogenetics data sets on the ASD cohort and CGH data on an additional 1000 control samples. -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 RESEARCH ARTICLE A Mutation in Histone H2B Represents a New Class of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,4, Rajarshi P. Ghosh5, Adrian T. Grzybowski6, Tao Yu7, Qiao Zhang8, Alberto Riva9, Tanmay P. Lele8, George C. Schatz4, Neil L. Kelleher4, Alexander J. Ruthenburg6, Jan Liphardt5, and Jonathan D. Licht1 Downloaded from cancerdiscovery.aacrjournals.org on September 30, 2021. © 2019 American Association for Cancer Research. Published OnlineFirst July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 ABSTRACT By examination of the cancer genomics database, we identified a new set of mutations in core histones that frequently recur in cancer patient samples and are predicted to disrupt nucleosome stability. In support of this idea, we characterized a glutamate to lysine mutation of histone H2B at amino acid 76 (H2B-E76K), found particularly in bladder and head and neck cancers, that disrupts the interaction between H2B and H4. Although H2B-E76K forms dimers with H2A, it does not form stable histone octamers with H3 and H4 in vitro, and when recon- stituted with DNA forms unstable nucleosomes with increased sensitivity to nuclease. Expression of the equivalent H2B mutant in yeast restricted growth at high temperature and led to defective nucleosome-mediated gene repression. Significantly, H2B-E76K expression in the normal mammary epithelial cell line MCF10A increased cellular proliferation, cooperated with mutant PIK3CA to pro- mote colony formation, and caused a significant drift in gene expression and fundamental changes in chromatin accessibility, particularly at gene regulatory elements. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7