Catalyzed Reaction of Isocyanates (RNCO) with Water
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Use and Storage of Methyl Isocyanate (MIC) at Bayer Cropscience
Summary The Use and Storage of Methyl Isocyanate (MIC) at Bayer CropScience The use of hazardous chemicals such as methyl isocyanate can be a significant concern to the residents of communities adjacent to chemical facilities, but is often an integral, necessary part of the chemical manufacturing process. In order to ensure that chemical manufacturing takes place in a manner that is safe for workers, members of the local community, and the environment, the philosophy of inherently safer processing can be used to identify opportuni ties to eliminate or reduce the hazards associated with chemical processing. However, the concepts of inherently safer process analysis have not yet been adopted in all chemical manu facturing plants. This report presents a possible framework to help plant managers choose between alternative processing options—considering factors such as environmental impact and product yield as well as safety—to develop a chemical manufacturing system. n 2008, an explosion at the Bayer CropScience chemical production Iplant in Institute, West Virginia, resulted in the deaths of two employees, a fire within the production unit, and extensive damage to nearby structures. The accident drew renewed attention to the fact that the Bayer facility manufac tured and stored methyl isocyanate, or MIC—a volatile, highly toxic chemical used in the production of carbamate pesticides and the agent responsible for thousands of deaths in Bhopal, India, in 1984. In the Institute incident, debris Figure 1. The Bayer CropScience facility at Insitute, WV. from the blast hit the shield surrounding Google Earth satellite image: © 2012 Google a MIC storage tank, and although the container was not damaged, an investiga tion by the U.S. -

Assessment of Portable HAZMAT Sensors for First Responders
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Assessment of Portable HAZMAT Sensors for First Responders Author(s): Chad Huffman, Ph.D., Lars Ericson, Ph.D. Document No.: 246708 Date Received: May 2014 Award Number: 2010-IJ-CX-K024 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant report available electronically. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Assessment of Portable HAZMAT Sensors for First Responders DOJ Office of Justice Programs National Institute of Justice Sensor, Surveillance, and Biometric Technologies (SSBT) Center of Excellence (CoE) March 1, 2012 Submitted by ManTech Advanced Systems International 1000 Technology Drive, Suite 3310 Fairmont, West Virginia 26554 Telephone: (304) 368-4120 Fax: (304) 366-8096 Dr. Chad Huffman, Senior Scientist Dr. Lars Ericson, Director UNCLASSIFIED This project was supported by Award No. 2010-IJ-CX-K024, awarded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect those of the Department of Justice. This document is a research report submitted to the U.S. Department of Justice. This report has not been published by the Department. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -

What Is in Your Cigarette?
What Is In Your Cigarette? There are more than 4,000 ingredients, including 43 known cancer-causing (carcinogenic) compounds and 400 other toxins in a cigarette other than tobacco. Common additives include yeast, wine, caffeine, beeswax and chocolate. Here are some other ingredients: • Ammonia • Cadmium • Megastigmatrienone (Household cleaner) (Used in batteries) (Chemical naturally found in grapefruit juice) • Angelica root extract • Cyanide (Deadly poison) (Known to cause cancer in • Maltitol • DDT (A banned insecticide) animals) (Sweetener for diabetics) • Ethyl Furoate • Arsenic • Napthalene (Causes liver damage in (Used in rat poisons) (Ingredient in mothballs) animals) • Benzene • Methyl isocyanate • Lead (Used in making dyes, (Its accidental release killed (Poisonous in high doses) synthetic rubber) 2000 people in Bhopal, India in 1984) • Formaldehiyde (Used to • Butane preserve dead specimens) (Gas; used in lighter fluid) • Polonium (Cancer-causing radioactive • Methoprene (Insecticide) • Carbon monoxide element) (Poisonous gas) Important to know: • Fungicides and pesticides -- Cause many types of cancers and birth defects. • Cadmium -- Linked to lung and prostate cancer. • Benzene -- Linked to leukemia. • Formaldehyde -- Linked to lung cancer. • Nickel -- Causes increased susceptibility to lung infections. If you are angry that so many things have been added to the cigarettes you enjoy so much, you should be. Many of these chemicals were added to make you better able to tolerate toxic amounts of cigarette smoke. Make sure that you have the last laugh. Regardless of the countless chemicals in your cigarettes, quitting is always your option. Cigna Members: Call (855) 246 – 1873 to speak with a health coach today! Non-Cigna Members: Call (877) U – Can – Now for the Florida Quit Line to speak to a health coach today! . -

1. Detta Föreställer Kapitelrubrik (1K Kapitelrubrik)
SCOEL/SUM/118 September 2006 Recommendation from the Scientific Committee on Occupational Exposure Limits for Methyl Isocyanate 8-hour TWA: - STEL (15 min): 0,02 ppm Additional classification: - Substance Identity and Properties CAS No.: 624-83-9 Synonyms: isocyanic acid methylester Structure: H3C-N=C=O Molecular weight: 57.06 Boiling point: 39°C Melting point: -45°C Vapour pressure: 46.4 kPa (20°C) Conversion factors: 1 ppm = 2.4 mg/m3 1 mg/m3 = 0.4 ppm Classification: F+; R12 Extremely flammable Repr.Cat.3; R63 Possible risk of harm to the unborn child T+; R26 Very toxic by inhalation T; R24/25 Toxic in contact with skin and if swallowed R42/43 May cause sensitization by inhalation and skin contact Xi; R37/38 Irritating to respiratory system and skin R41 Risk of serious damage to eyes 1 This summary document is based on a consensus document from the Swedish Criteria Group for Occupational Standards (Montelius, 2002). Methyl isocyanate (MIC) is a monoisocyanate and should be distinguished from the diisocyanates. At room temperature MIC is a clear liquid. It is sparingly soluble in water, although on contact with water it reacts violently, producing a large amount of heat. MIC has a sharp odour with an odour threshold above 2 ppm (Römpp and Falbe, 1997). Occurrence and Use Methylisocyanate (MIC) occurs primarily as an intermediate in the production of carbamate pesticides. It has also been used in the production of polymers (Hrhyhorczuk et al., 1992). Photolytic breakdown of N-methyldithiocarbamate releases some MIC, and it can therefore occur in the air around application of the pesticides (Geddes et al., 1995). -

Methyl Isocyanate (Mic)
METHYL ISOCYANATE (MIC) I. Protocol Overview Methyl isocyanate, classified as a choking/pulmonary agent, is a water reactive liquid at room temperature. Methyl isocyanate is irritating and corrosive to the eyes, skin, and respiratory tract, including asthma-like allergy. Exposure to methyl isocyanate can result in cough, difficulty breathing, chest pain, excessive tearing (lacrimation), swelling of the eyes, and unconsciousness. Late stage exposure (next 24 to72 hours) can include acute lung injury, cardiac arrest, and death. Decontamination of patients is absolutely critical. For all suspected chemical exposures, consult the Poison Control Center (800-222-1222) located at Children’s Hospital of Philadelphia. Information and treatment advice is available to the public and healthcare professionals at no charge. Methyl isocyanate is an intermediate chemical in the production of carbamate pesticides (such as carbaryl, carbofuran, methomyl, and aldicarb). It has been used in the production of rubbers and adhesives. The Bhopal India Disaster of 1984 was caused by the accidental release of 40 tons of methyl isocyanate (MIC) gas from a Union Carbide pesticide plant. Greenpeace cites 20,000 total deaths from related illnesses as its conservative estimate with nearly 3,000 people dead initially. Bhopal is frequently cited as the world's worst industrial disaster. There are no clinical biomarkers known for methyl isocyanate exposure. Routine laboratory studies include chest radiography and pulse oximetry (or ABG measurements). Rapid toxicological screening may rule out other exposures. Air samples may be analyzed using XAD-7 adsorption tubes followed analysis by high performance liquid chromatography (HPLC) using a fluorescence or ultraviolet (UV) detector. The Delaware Public Health Laboratory does not perform this testing. -
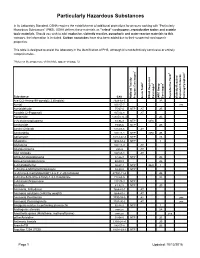
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Air Target Concentration: 50 Μg/M3 (20 Ppb) (OSHA PEL) Procedure
METHYL ISOCYANATE (MIC) Method no.: 54 Matrix: Air Target concentration: 50 µg/m3 (20 ppb) (OSHA PEL) Procedure: Samples are collected by drawing a known volume of air through XAD-7 tubes coated with 0.3 mg of 1-(2-pyridyl)piperazine (1-2PP). Samples are desorbed with acetonitrile (ACN) and analyzed by high performance liquid chromatography (HPLC) using a fluorescence or ultraviolet (UV) detector. Recommended air volume and sampling rate: 15 L at 0.05 L/min Reliable quantitation limit: 4.8 µg/m3 (1.9 ppb) Standard error of estimate at the target concentration: 8.0% (Section 4.5.) Special requirements: The coated XAD-7 tubes should be stored under refrigeration before sampling. Status of method: Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. Date: April 1985 Chemists: Donald Burright Duane Lee Carcinogen and Pesticide Branch OSHA Analytical Laboratory Salt Lake City, Utah 1 of 13 T-54-FV-01-8504-M 1. General Discussion 1.1. Background 1.1.1. History In the past there has been no validated sampling and analytical procedures for methyl isocyanate (MIC) in air that could be used for compliance purposes. A search of the literature produced three procedures that were candidates for a validated method: The first procedure contained only analytical conditions for MIC derivatized with Nitro Reagent (nitrobenzyl-N-n-propylamine). (Ref. 5.1.) No data were presented to indicate the collection efficiency of the sampling device. The second procedure was a failure report by NIOSH that had derivatized MIC with Nitro Reagent or NMA (naphthylmethylamine) in a toluene impinger. -

Pesticides and Intermediates, Supp. A
PROCESS ECONOMICS PROGRAM SRI INTERNATIONAL Menlo Park, California ABSTRACT 94025 l Process Economics Program Report No. 171A (August 1985) -0 This report describes and evaluates the processes for making carbo- furan and mancozeb, as well as the intermediates for these two pesti- cides. Carbofuran is made from either catechol or o-nitrochlorobenzene. The economics of each route, including the manufacture of the inter- mediates methyl isocyanate and methallyl chloride, are evaluated. The catechol route is found to be economically superior. For methyl iso- cyanate, which is common to both routes, two nonphosgenation processes are evaluated and compared with two phosgenation processes. The non- phosgenation route is not only competitive with the phosgenation route, but also avoids the transport of both methyl isocyanate and Phosgene when integrated with carbofuran production. Mancoxeb is evaluated in detail, with a brief evaluation of two similar pesticides, maneb, and zineb. The intermediates are carbon disulfide and ethylenediamine. The process for making carbon disulfide from methane and sulfur is evaluated, with a discussion of the use of alternative carbon sources. For ethylenediamine, two processes start- ing from ethylene dichloride, one process from monoethanolamine, and 0 another from ethylene oxide are evaluated and compared. PEP'84 YCY, DJL, CSL - Report No.171A 0 PESTICIDES AND INTERMEDIATES SUPPLEMENT A by YEN-CHEN YEN with contributions by DIN-JIN LIN 0 and CHUN-SAN LIU August 1985 A private report by the PROCESS ECONOMICS PROGRAM Menlo Park, California 94025 For detailed marketing data and information, the reader is referred to one of the SRI programs specializing in marketing research. -

Spray Foam Event 2013, Session a Presentations Contents List
Spray Foam Event 2013, Session A Presentations Contents List 1. Introduction to Spray Polyurethane Foam (Click to go to PDF Page 2) 2. OSHA’s Isocyanates National Emphasis Program (Click to go to PDF Page 58) 3. EOLWD On-site Consultation Program (Click to go to PDF Page 76) 4. Essential Resources and Training, American Chemistry Council (Click to go to PDF Page 109) 5. Safe Spray Foam (Click to go to PDF Page 126) 6. EPA Safe Use of Polyurethane Products (Click to go to PDF Page 137) Return to Contents List Introduction to Spray Polyurethane Foam This presentation will provide important background information on SPF, including history, product categories and delivery methods and applications. It will also address chemical concerns and include tips for a quality installation, and briefly cover environmental impacts of the product COPYRIGHTED MATERIALS This presentation is protected by US and International copyright laws. Reproduction, distribution, display and use of any part of this presentation without written permission of the speaker is prohibited. © 2013 Spray Polyurethane Foam Alliance Presentation Content 1. History 2. Product Categories 3. Basic Chemistry 4. Delivery Methods 5. Chemical Concerns 6. Environmental Impact 7. Quality Installation 8. Summary History of SPF in Buildings in construction for 50 years • Late 60’s ‐ Medium Density (agricultural and industrial) • Mid 70’s ‐Roofing ‐ Medium Density (general const.) ‐ Sealants • Mid 90’s ‐ Low Density (residential) Product Category Four general categories of SPF Spray Foam -
![[Cas No. 624-83-9]](https://docslib.b-cdn.net/cover/9373/cas-no-624-83-9-1639373.webp)
[Cas No. 624-83-9]
External Draft March 2015 1 2 3 4 5 6 7 IMMEDIATELY DANGEROUS TO LIFE OR HEALTH (IDLH) VALUE PROFILE 8 9 10 11 FOR 12 13 14 15 METHYL ISOCYANATE 16 17 18 19 [CAS NO. 624-83-9] 20 21 22 23 24 25 26 Department of Health and Human Services 27 Centers for Disease Control and Prevention 28 National Institute for Occupational Safety and Health 29 30 This information is distributed solely for the purpose of pre dissemination peer review under applicable information quality guidelines. It has not been formally disseminated by the National Institute for Occupational Safety and Health. It does not represent and should not be construed to represent any agency determination or policy. i External Draft March 2015 1 Disclaimer 2 3 Mention of any company or product does not constitute endorsement by the National Institute for Occupational 4 Safety and Health (NIOSH). In addition, citations to Web sites external to NIOSH do not constitute NIOSH 5 endorsement of the sponsoring organizations or their programs or products. Furthermore, NIOSH is not 6 responsible for the content of these Web sites. 7 8 Ordering Information 9 10 To receive this document or information about other occupational safety and health topics, contact NIOSH: 11 12 Telephone: 1-800-CDC-INFO (1-800-232-4636) 13 TTY: 1-888-232-6348 14 E-mail: [email protected] 15 Or visit the NIOSH Web site: www.cdc.gov/niosh 16 17 For a monthly update on news at NIOSH, subscribe to NIOSH eNews by visiting www.cdc.gov/niosh/eNews. -

Methyl Isocyanate Mis
METHYL ISOCYANATE MIS CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: 7.1 Grades of Purity: Commercial (99%) Common Synonyms Liquid Colorless Sharp, unpleasant Currently not available 7.2 Storage Temperature: It is recommended that odor Isocyanatomethane 4.2 Flammable Limits in Air: 5.3% - 26% bulk quantities be cooled to approximately 0°C. Isocyanic acid, methyl ester 4.3 Fire Extinguishing Agents: Small fires: Drums may be stored at ambient temperature Methane, isocyanato- out of direct sunlight. Storage temperature Methyl carbonimide Floats; slowly mixes and slowly reacts with water at 20°C. dry chemical, CO2, water spray or foam; should not exceed 30°C. MIC large fires: water spray, fog or foam. 4.4 Fire Extinguishing Agents Not to Be 7.3 Inert Atmosphere: Must be protected by a dry Used: Not pertinent nitrogen (dew point -40°C. or lower) KEEP PEOPLE AWAY. AVOID CONTACT WITH LIQUID AND VAPOR. atmosphere. EVACUATE AREA. 4.5 Special Hazards of Combustion Wear chemical protective suit with self-contained breathing apparatus. Products: Contain toxic and irritating 7.4 Venting: Not listed Shut off ignition sources and call fire department. gases, including HCN and NOx. 7.5 IMO Pollution Category: Currently not available Stay upwind and use water spray to ``knock down'' vapor. 4.6 Behavior in Fire: Very flammable; may be 7.6 Ship Type: Currently not available Notify local health and pollution control agencies. ignited by heat, sparks or flames. May travel to a source of ignition and 7.7 Barge Hull Type: Currently not available Fire FLAMMABLE. -

14 Chemical Modification
Chemical Modification 14 of Wood Roger M. Rowell USDA, Forest Service, Forest Products Laboratory, and Department of Biological Systems Engineering, University of Wisconsin, Madison, WI CONTENTS 14.1 Degradation of Wood .........................................................................................................383 14.2 Chemical Reaction Systems...............................................................................................383 14.3 Chemical Reactions with Wood.........................................................................................385 14.3.1 Acetylation...........................................................................................................386 14.3.2 Acid Chlorides .....................................................................................................387 14.3.3 Other Anhydrides.................................................................................................387 14.3.4 Carboxylic Acids .................................................................................................388 14.3.5 Isocyanates ...........................................................................................................388 14.3.6 Formaldehyde ......................................................................................................388 14.3.7 Other Aldehydes ..................................................................................................389 14.3.8 Methylation ..........................................................................................................389