St-Onge Carle Myriam.Pdf (3.057Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Recent Syntheses of Steroidal Oxazoles, Oxazolines and Oxazolidines
A Platinum Open Access Journal Review for Organic Chemistry Free to Authors and Readers DOAJ Seal Arkivoc 2021, part i, 471-490 Recent syntheses of steroidal oxazoles, oxazolines and oxazolidines Besma Bendif,a,b Malika Ibrahim-Ouali,*a and Frédéric Dumur c aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, F-13397 Marseille, France bLaboratoire de Chimie Appliquée, Faculté des Sciences, Université du 08 mai 1945 Guelma, Algeria cAix Marseille Univ, CNRS, ICR, UMR 72 73, F-13397 Marseille, France Email: [email protected] Received 03-15-2021 Accepted 04-11-2021 Published on line 05-08-2021 Abstract It was found that the introduction of heterocycles to steroids often leads in a change of their physiological activity and the appearance of new interesting biological precursors. Recent developments in the syntheses of steroidal oxazoles, oxazolines, and oxazolidines are described herein. The biological activities of those steroidal derivatives for which data are available are given. Keywords: Steroids, oxazoles, oxazolines, oxazolidines DOI: https://doi.org/10.24820/ark.5550190.p011.512 Page 471 ©AUTHOR(S) Arkivoc 2021, i, 471-490 Bendif, B. et al. Table of Contents 1. Introduction 2. Synthesis of Steroidal Oxazoles 3. Synthesis of Steroidal Oxazolines 4. Synthesis of Steroidal Oxazolidines 5. Conclusions Acknowledgements References 1. Introduction Steroids constitute an extensive and important class of biologically active polycyclic compounds that are widely used for therapeutic purposes.1-3 Even after decades of research, the total synthesis of steroid nuclei by improved strategies continues to receive considerable attention. Numerous methods have been exploited for the total synthesis of steroids which are widely distributed in nature and which possess practical medical importance. -

A COORDINATION and REDOX CHEMISTRY of ANTIMONY-BASED LIGANDS and APPLICATIONS in ORGANOMETALLIC CATALYSIS a Dissertation by YING
COORDINATION AND REDOX CHEMISTRY OF ANTIMONY-BASED LIGANDS AND APPLICATIONS IN ORGANOMETALLIC CATALYSIS A Dissertation by YING-HAO LO Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, François P. Gabbaï Committee Members, Michael B. Hall Michael Nippe Hung-Jen Wu Head of Department, Simon W. North December 2019 Major Subject: Chemistry Copyright 2019 Ying-Hao Lo a ABSTRACT The use of Z-ligands to modulate the electronic property of transition metal centers is a powerful strategy in catalyst design. Our group has shown that antimony- based ambiphilic ligand, accept electron density from adjacent metal centers by the σ* orbital of the antimony (V) center and thus increasing the electrophilic reactivity of the trancition metal center. In this thesis, we were eager to determine if the charge of the Z- type ligand can be used to further enhance this effect. To this end, we synthesized a 2+ 2+ dicationic gold complex [46] ([(o‐(Ph2P)C6H4)2(o‐Ph2PO)C6H4)SbAuCl] ) featuring a dicationic antimony (V) ligand with a phosphine oxide arm coordinating to the antimony center. Both experimental and computational results show that the gold complex possess a strong Au→Sb interaction reinforced by the dicationic character of the antimony center. The gold‐bound chloride anion of this complex is rather inert and necessitates the addition of excess AgNTf2 to undergo activation. The activated complex, referred to as 2+ 2+ [47] ([((o‐(Ph2P)C6H4)2(o‐Ph2PO)C6H4)SbAuNTf2] ) readily catalyzes both the polymerization and the hydroamination of styrene. -

DEVELOPMENT of NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS by LINDSEY RAE CULLEN DISSERTATION Submitted
DEVELOPMENT OF NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS BY LINDSEY RAE CULLEN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Scott E. Denmark, Chair Professor Martin D. Burke Professor Scott K. Silverman Professor Steven C. Zimmerman ii Abstract The first half of this thesis (Chapter 2) described the development of a fluoride-promoted conjugate addition of sulfur-stabilized carbanion nucleophiles to α,β-unsaturated ketones and esters. This reaction was achieved using a substoichiometric amount of TBAF, resulting in high yields on the desired 1,4-addition product. The addition of 1,3-dithianes was given particular focus as a novel method for the preparation of differentially protect 1,4-dicarbonyl compounds. Observation by 13C NMR spectroscopy provided evidence that the reaction proceeds through an ion pair, and attempts to extend this reaction to asymmetric additions using a chiral counterion are presented in detail. The second half of this thesis (Chapter 3) details development of a phase transfer catalyzed [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds. Initial investigation focused on identifying viable substrate classes that would undergo selective [2,3]-rearrangement under phase transfer conditions. Under certain conditions, the [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds takes place in the presence of a phase transfer agent, providing a rare example of a phase transfer catalyzed unimolecular reaction. In the course of this investigation it was found that catalysis is dependent on several variables including base concentration, catalyst structure, and substrate lipophilicity. -

Argonne Report.Pdf
CONTENTS NOTATION ........................................................................................................................... xi ABSTRACT ........................................................................................................................... 1 1 INTRODUCTION ........................................................................................................... 5 1.1 Overview of the Emergency Response Guidebook ................................................ 5 1.2 Organization of this Report ..................................................................................... 7 2 GENERAL METHODOLOGY ....................................................................................... 9 2.1 TIH List ................................................................................................................... 10 2.1.1 Background ................................................................................................. 10 2.1.2 Changes in the TIH List for the ERG2012 ................................................. 11 2.2 Shipment and Release Scenarios ............................................................................ 11 2.2.1 Shipment Profiles ........................................................................................ 12 2.2.2 Treatment of Chemical Agents ................................................................... 14 2.3 Generics, Mixtures, and Solutions .......................................................................... 17 2.4 Analysis of Water-Reactive -

IODINE Its Properties and Technical Applications
IODINE Its Properties and Technical Applications CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York IODINE Its Properties and Technical Applications ¡¡iiHiüíiüüiütitittüHiiUitítHiiiittiíU CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York 1951 Copyright, 1951, by Chilean Iodine Educational Bureau, Inc. Printed in U.S.A. Contents Page Foreword v I—Chemistry of Iodine and Its Compounds 1 A Short History of Iodine 1 The Occurrence and Production of Iodine ....... 3 The Properties of Iodine 4 Solid Iodine 4 Liquid Iodine 5 Iodine Vapor and Gas 6 Chemical Properties 6 Inorganic Compounds of Iodine 8 Compounds of Electropositive Iodine 8 Compounds with Other Halogens 8 The Polyhalides 9 Hydrogen Iodide 1,0 Inorganic Iodides 10 Physical Properties 10 Chemical Properties 12 Complex Iodides .13 The Oxides of Iodine . 14 Iodic Acid and the Iodates 15 Periodic Acid and the Periodates 15 Reactions of Iodine and Its Inorganic Compounds With Organic Compounds 17 Iodine . 17 Iodine Halides 18 Hydrogen Iodide 19 Inorganic Iodides 19 Periodic and Iodic Acids 21 The Organic Iodo Compounds 22 Organic Compounds of Polyvalent Iodine 25 The lodoso Compounds 25 The Iodoxy Compounds 26 The Iodyl Compounds 26 The Iodonium Salts 27 Heterocyclic Iodine Compounds 30 Bibliography 31 II—Applications of Iodine and Its Compounds 35 Iodine in Organic Chemistry 35 Iodine and Its Compounds at Catalysts 35 Exchange Catalysis 35 Halogenation 38 Isomerization 38 Dehydration 39 III Page Acylation 41 Carbón Monoxide (and Nitric Oxide) Additions ... 42 Reactions with Oxygen 42 Homogeneous Pyrolysis 43 Iodine as an Inhibitor 44 Other Applications 44 Iodine and Its Compounds as Process Reagents ... -

Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates
Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates by Alexandria Daria Maria Jeanneret A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Alexandria Daria Maria Jeanneret 2018 Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalates Alexandria Daria Maria Jeanneret Master of Science Department of Chemistry University of Toronto 2018 Abstract Nitriles are considered very versatile functional groups due to their ability to easily be transformed into a variety of other functional groups in one or two steps. In particular, the synthesis of α-arylnitriles is of interest to organic chemists due to their presence in pharmaceuticals and their value as synthetic intermediates. Taking advantage of nickel’s unique ability to insert into a C‒O bond, the focus of this thesis is on the nickel-catalyzed cyanation of benzylic and allylic pivalates, exploring the use of inorganic and organic cyanide sources for this transformation. The substrate scope for the synthesis of benzylic and allylic nitriles will be presented as well as studies examining the functional group tolerance of this cyanation reaction, which led to further insights into the mechanism and applicability of this chemistry. ii Acknowledgments First and foremost, I would like to thank Professor Sophie Rousseaux for the opportunity to work to work in her lab over the last year. Her passion for chemistry is beyond contagious and her guidance has been invaluable. I would also like to thank Professor Mark Taylor for his help with this thesis. Secondly, I would like to thank all the staff at the NMR and AIMS facility for all their hard-work and dedication to ensuring the instruments run smoothly and for always taking the time to answer questions. -
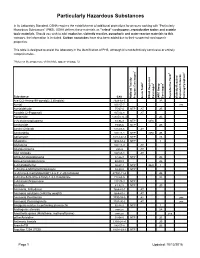
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Interhalogen Compounds
INTERHALOGEN COMPOUNDS Smt. EDNA RICHARD Asst. Professor Department of Chemistry INTERHALOGEN COMPOUND An interhalogen compound is a molecule which contains two or more different halogen atoms (fluorine, chlorine, bromine, iodine, or astatine) and no atoms of elements from any other group. Most interhalogen compounds known are binary (composed of only two distinct elements) The common interhalogen compounds include Chlorine monofluoride, bromine trifluoride, iodine pentafluoride, iodine heptafluoride, etc Interhalogen compounds into four types, depending on the number of atoms in the particle. They are as follows: XY XY3 XY5 XY7 X is the bigger (or) less electronegative halogen. Y represents the smaller (or) more electronegative halogen. Properties of Interhalogen Compounds •We can find Interhalogen compounds in vapour, solid or fluid state. • A lot of these compounds are unstable solids or fluids at 298K. A few other compounds are gases as well. As an example, chlorine monofluoride is a gas. On the other hand, bromine trifluoride and iodine trifluoride are solid and liquid respectively. •These compounds are covalent in nature. •These interhalogen compounds are diamagnetic in nature. This is because they have bond pairs and lone pairs. •Interhalogen compounds are very reactive. One exception to this is fluorine. This is because the A-X bond in interhalogens is much weaker than the X-X bond in halogens, except for the F-F bond. •We can use the VSEPR theory to explain the unique structure of these interhalogens. In chlorine trifluoride, the central atom is that of chlorine. It has seven electrons in its outermost valence shell. Three of these electrons form three bond pairs with three fluorine molecules leaving four electrons. -

Ghs Reference Materials Health Hazard Criteria
APPENDIX F: GHS REFERENCE MATERIALS This appendix provides both an overview of GHS highly toxic hazard classification. A listing of Particularly Hazardous Substances that Carnegie Mellon University has published with their Chemical Hygiene plan is also provided as general reference. The list is useful cross check with GHS listings to determine which materials require prior approval for use BUT NO LIST IS COMPLETE you must check the SDS for possible additional chemicals rated as highly toxic. This appendix also provides the GHS (global harmonization system) for chemical hazard classification under the Hazard Communication Standard for highly toxic materials. This section provides overall information about categories under the classification of acute toxicity, mutagens’, reproductive and carcinogen hazards. When chemicals are rated on the GHS – Safety Data Sheet (SDS) as the following hazards then the PRIOR APPROVAL PROCESS WITH CHEMICAL HYGIENE OFFICER/COMMITTEE must be used: Acute toxicity category 1 and 2, Germ cell mutagenicity as a category 1A Substances known to induce heritable mutations in germ cells of humans and Category 1B: Substances which should be regarded as if they induce heritable mutations in the germ cells of humans, Reproductive Hazard as a category 1: Known or presumed human reproductive toxicants and Category 2; suspected human reproductive toxicant. Carcinogen as a Category 1 (includes 1A and 1B): Known or presumed human carcinogens, Category 2: Suspected human carcinogens. The Campus Chemical Hygiene Committee (Officer) must conduct a prior approval process. Appendix C Chemical Prior Approval Form on procedure for conducing prior approval. The following is from OSHA standard on the chemicals classifications that PCC Laboratory instructional operations shall use for defining the prior approval hazards. -

United States Patent Office Patented Mar
3,171,249 United States Patent Office Patented Mar. 2, 1965 i 2 fuel rocket engine. The above and other objects of this 3,171,249 PROPELLANT AND Rick |PROPULSSON METH invention will become apparent from the discussion OXD EMPLOYANG EYERAZINE WITH AMSNO which follows. TETRAZOLES The objects of this invention are accomplished by the Ronald E. Be, Canoga Park, Calif., assigner to use of compounds having the general formula: North American Aviatiosa, Bac. No Drawing. Fied Nov. 29, 1961, Ser. No. 155,803 NH2. (R) 8 Clains. (C. 60-35.4) N This invention relates to a novel rocket propellant. YS More particularly, this invention relates to a novel in 10 N--H proved rocket propellant and a method of operating a wherein x varies from 0 to 1 and R is selected from the rocket engine. class consisting of HCl, H2O, HNO3, and HCIO, as addi The criterion by which rocket propellants are classi tives to a hydrazine-based rocket fuel in an amount suffi fied is specific impulse, Is, defined as thrust in pounds 5 cient to depress the freezing point at least 40° C. while divided by the total mass flow of fuel and oxidizer in retaining about the same density impulse and specific pounds per second. Specific impulse is thus given in impulse. Hence, an embodiment of this invention com units of "seconds.” Oxidizer-fuel propulsion system prises a method of operating a rocket engine comprising compositions with a relatively high specific impulse are ejecting from the reaction chamber of the engine a gaseous known in the art. -

How to Identify and Report Hazardous Substances on The
OREGON COMMUNITY RIGHT TO KNOW AND PROTECTION ACT How to Identify and Report Hazardous Substances on the Hazardous Substance Information Survey February 2012 For assistance call the Mailing Address: Hazardous Substance Office of State Fire Marshal Information Hotline Community Right to Know Unit 4760 Portland Rd NE (503) 378-6835 Salem, OR 97305-1760 Toll Free (800) 454-6125 TDD (503) 390-4661 Monday – Friday Website: 8AM – 12PM and 1PM – 5PM http://www.oregon.gov/OSP/SFM/CR2K_Home.shtml Visit our website for more information: http://www.oregon.gov/OSP/SFM/CR2K_Home.shtml. These documents are currently available from our website: Blank Section D Chemical Form Blank Section E Additional Storage Location Form Survey Request Form Gas Conversion Chart Survey Mailing Schedule TABLE OF CONTENTS Introduction…………………………………………………………….. 1 Quick Steps to Complete the Survey …………………………………. 1 Reporting Requirements ……………………………...................... 2 What is a Hazardous Substance? ……………………………………... 3 What is a Reportable Quantity? ……………………………………….3 Reporting Compressed Gases ……………………………………….. 4 Liquefied and Cryogenic Gases …………………………………........ 4 Reporting Lead Acid Batteries ……………………………………….. 5 Tables for Completing the Survey ……………………………………... 6 Instructions and Definitions ……………………………………………. 7 Reporting Storage Locations …………………………………………..11 Frequently Asked Questions ………………………………………….. 13 EHS, 112r, PSM Questions …………………………………………… 14 EHS List ..............................................................................15 112(r) List ............................................................................18