Rapid Screening of UPB1 Gene Variations by High Resolution Melting Curve Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Dihydropyrimidine Dehydrogenase in the Metabolism of the Anticancer Drugs
Cancer Chemotherapy and Pharmacology (2019) 84:1157–1166 https://doi.org/10.1007/s00280-019-03936-w REVIEW ARTICLE Dihydropyrimidine dehydrogenase in the metabolism of the anticancer drugs Vinay Sharma1 · Sonu Kumar Gupta1 · Malkhey Verma1 Received: 3 May 2019 / Accepted: 21 August 2019 / Published online: 4 September 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Cancer caused by fundamental defects in cell cycle regulation leads to uncontrolled growth of cells. In spite of the treatment with chemotherapeutic agents of varying nature, multiple resistance mechanisms are identifed in cancer cells. Similarly, numerous variations, which decrease the metabolism of chemotherapeutics agents and thereby increasing the toxicity of anticancer drugs have been identifed. 5-Fluorouracil (5-FU) is an anticancer drug widely used to treat many cancers in the human body. Its broad targeting range is based upon its capacity to act as a uracil analogue, thereby disrupting RNA and DNA synthesis. Dihydropyrimidine dehydrogenase (DPD) is an enzyme majorly involved in the metabolism of pyrimidines in the human body and has the same metabolising efect on 5-FU, a pyrimidine analogue. Multiple mutations in the DPD gene have been linked to 5-FU toxicity and inadequate dosages. DPD inhibitors have also been used to inhibit excessive degradation of 5-FU for meeting appropriate dosage requirements. This article focusses on the role of dihydropyrimidine dehydrogenase in the metabolism of the anticancer drug 5-FU and other associated drugs. Keywords Cancer · Anticancer drugs · Dihydropyrimidine dehydrogenase (DPD) · 5-Fluorouracil (5-FU) · Drug resistance · Drug metabolism Introduction by Dihydropyrimidine dehydrogenase (DPD) through the pyrimidine degradation pathway. -
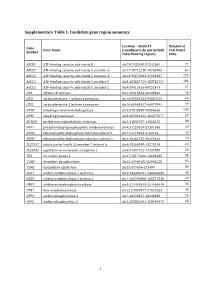
Supplementary Table 1. Candidate Gene Region Summary
Supplementary Table 1. Candidate gene region summary Location – Build 37 Number of Gene Gene Name (coordinates do not include Test Panel Symbol 25kb flanking region) SNPs ABCB1 ATP-binding cassette, sub-family B chr7:87132948-87342564 77 ABCC3 ATP-binding cassette, sub-family C, member 3 chr17:48712218-48769063 64 ABCC4 ATP-binding cassette, sub-family C, member 4 chr13:95672083-95953687 224 ABCC5 ATP-binding cassette, sub-family C, member 5 chr3:183637724-183735727 101 ABCG2 ATP-binding cassette, sub-family G, member 2 chr4:89011416-89152474 57 CDA cytidine deaminase chr1:20915444-20945400 26 CES1 carboxylesterase 1 isoform a precursor chr16:55836764-55867075 24 CES2 carboxylesterase 2 isoform a precursor chr16:66968347-66978994 59 DPYD dihydropyrimidine dehydrogenase chr1:97543300-98386615 239 DPYS dihydropyrimidinase chr8:105391652-105479277 69 MTHFR methylenetetrahydrofolate reductase chr1:11845787-11866115 38 PPAT phosphoribosyl pyrophosphate amidotransferase chr4:57259529-57301845 29 RRM1 ribonucleoside-diphosphate reductase subunit 1 chr11:4115924-4160106 29 RRM2 ribonucleoside-diphosphate reductase subunit 2 chr2:10262735-10270623 19 SLC22A7 solute carrier family 22 member 7 isoform b chr6:43265998-43273276 26 SLC29A1 equilibrative nucleoside transporter 1 chr6:44187242-44201888 26 TK1 thymidine kinase 1 chr17:76170160-76183285 35 TYMP thymidine phosphorylase chr22:50964182-50968258 92 TYMS thymidylate synthetase chr18:657604-673499 34 UCK1 uridine-cytidine kinase 1 isoform a chr9:134399191-134406655 43 UCK2 uridine-cytidine kinase 2 isoform a chr1:165796890-165877339 22 UMPS uridine monophosphate synthase chr3:124449213-124464040 34 UPB1 beta-ureidopropionase chr22:24890077-24922553 30 UPP1 uridine phosphorylase 1 chr7:48128355-48148330 16 UPP2 uridine phosphorylase 2 chr2:158851691-158992478 43 1 Supplementary Table 2. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2. -

Hypoxia Drives Dihydropyrimidine Dehydrogenase Expression in Macrophages
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.15.341123; this version posted October 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Hypoxia Drives Dihydropyrimidine Dehydrogenase Expression in Macrophages 2 and Confers Chemoresistance in Colorectal Cancer 3 4 Marie Malier1,2, Magali Court1,2, Khaldoun Gharzeddine1,2,3, Marie-Hélène Laverierre1,2,4, Sabrina 5 Marsili5,6, Fabienne Thomas5,6, Thomas Decaens 2,7,8, Gael Roth 2,7,8, Arnaud Millet1,2,3 6 1. Team Mechanobiology, Immunity and Cancer, Institute for Advanced Biosciences Inserm 1209 – 7 UMR CNRS 5309, Grenoble, France 8 2. Grenoble Alpes University, Grenoble, France 9 3. Research department, University Hospital Grenoble-Alpes, Grenoble, France 10 4. Department of pathological anatomy and cytology, University Hospital Grenoble-Alpes, Grenoble, 11 France 12 5. CRCT Inserm U037, Toulouse University 3, Toulouse, France 13 6. Claudius Regaud Institute, IUCT-Oncopole, Toulouse, France 14 7. Department of Hepatogastroenterology, University Hospital Grenoble-Alpes, Grenoble, France 15 8. Team Tumor Molecular Pathology and Biomarkers, Institute for Advanced Biosciences UMR 16 Inserm 1209 – CNRS 5309, Grenoble, France 17 18 Corresponding author: 19 Arnaud Millet MD PhD 20 Team Mechanobiology, Immunity and Cancer, Institute for Advanced Biosciences 21 Batiment Jean Roget 3rd floor 22 Domaine de la Merci 23 38700 La Tronche 24 France 25 e-mail: [email protected] 26 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.15.341123; this version posted October 15, 2020. -

Novel and Rare Cnvs
Novel and rare CNVs 60.00 50.00 40.00 ge ta n e30.00 rc e P20.00 10.00 0.00 <10 kb 10-30 kb 30-100 kb >100 kb Size range Figure S1 Size distribution of the 946 novel and rare CNVs. A. Prasad et al. 1 SI A B 100564 MM0177-3 Figure S3 Results from ancestry analysis using SNP genotype data. (A) The figure shows the dimensions 1 and 2 of the multidimensional scaling. (B) A zoomed view of the Figure S2 (A) Genome browser view of 16p11.2 duplication (B) Genome browser view of 22q11.22-q11.23 duplication region 2 SI A. Prasad et al. A B A. Prasad et al. 3 SI C Figure S3 Results from ancestry analysis using SNP genotype data. (A) The figure shows the dimensions 1 and 2 of the multidimensional scaling. (B) A zoomed view of the known and putative European group of the dimensions 1 and 2 (C) Plot of dimensions 1 and 3 of the multidimensional scaling. ASD samples are colored as gray and HapMap3 samples are colored in different colors. Utah residents with ancestry from northern and western Europe (CEU) – light blue; Tuscany in Italy (TSI) – green; Japanese in Tokyo (JPT) – red; Han Chinese in Beijing (CHB) – yellow; Yoruba in Nigeria (YRI), Masai in Kenya (MKK), Luhya in Kenya (LWK)-black; African ancestry in Southwest USA (ASW)-dark blue; Gujarati Indians in Houston (GIH) – brown; Mexicans (MEX)- magenta 4 SI A. Prasad et al. Table S1 Samples with CNVs larger than 5 Mb in size Sample Sex Tissue Chr Size (bp) CNV Karyotype Other arrays detected by Affy500K, Marshall et 119975L M L 9p22.1-p21.3 4,998,116 loss n/a al. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Amidohydrolases of the Reductive Pyrimidine Catabolic Pathway Purification, Characterization, Structure, Reaction Mechanism
Frontiers in Drug, Chemistry and Clinical Research Review Article ISSN: 2631-5424 Amidohydrolases of the reductive pyrimidine catabolic pathway purification, characterization, structure, reaction mechanism and enzyme deficiency Schnackerz KD* Biocentre, University of Würzburg, Am Hubland, 97074 Würzburg, Germany Abstract In the reductive pyrimidine catalytic pathway uracil and thymine are converted to β- alanine and β-aminoisobutyrate. The amidohydrolases of this pathway are responsible for both the ring opening of dihydrouracil and dihydrothymine and the hydrolysis of N-carbamoyl-β-alanine and N-carbamoyl-β-aminoisobutyrate. The review summarize the properties, kinetic parameters, 3D-structures and reaction mechanisms of these proteins. The two amidohydrolases have unrelated folds, the DHPases belong to the amidohydrolase superfamily, while the βAS from higher eukaryotes belongs to the nitrilase superfamily. βAS from S. kluyveri is an exception to the rule and belongs to the Acyl/M20 family. Abbreviations: βAS: β-alanine synthase; HβAS: human β-alanine Amidohydolases of the pyrimidine degradative pathway synthase; NCβA: N-carbamoyl-β-alanine; DmβAS: β-alanine synthase from D. melanogaster; βAIBA: β-aminoisobutyrate; 5FU: 5-fluorouracil; Purification, properties, substrate specificities, and sequence DHPase: dihydropyrimidinase. homologies of dihydro-pyrimidinases In the catalytic pathway of pyrimidines, uracil and thymine are Dihydropyrimidinases (DHPases) from bovine, calf and rat liver [9- reduced to 5,6-dihydro-Uracil and 5,6-dihydrothymine, -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -

ß-Ureidopropionase Deficiency
Biochimica et Biophysica Acta 1822 (2012) 1096–1108 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis ß-Ureidopropionase deficiency: Phenotype, genotype and protein structural consequences in 16 patients André B.P. van Kuilenburg a,⁎, Doreen Dobritzsch b, Judith Meijer a, Michael Krumpel b, Laila A. Selim c, Mohamed S. Rashed d, Birgit Assmann e, Rutger Meinsma a, Bernhard Lohkamp b, Tetsuya Ito f, Nico G.G.M. Abeling a, Kayoko Saito g, Kaoru Eto h, Martin Smitka i, Martin Engvall j, Chunhua Zhang k, Wang Xu l, Lida Zoetekouw a, Raoul C.M. Hennekam m a Academic Medical Center, Emma Children's Hospital, Department of Clinical Chemistry, Laboratory Genetic Metabolic Diseases, 1105 AZ Amsterdam, The Netherlands b Karolinska Institutet, Dept. of Medical Biochemistry and Biophysics (MBB), S-17177 Stockholm, Sweden c Cairo University Children Hospital, Department of Pediatrics, Pediatric Neurology & Neurometabolic Divisions, Cairo, Egypt d Pharmagene Labs, Dokki 12311, Giza, Egypt e University Children's Hospital, Department of General Pediatrics, 40225 Düsseldorf, Germany f Nagoya City University, Graduate School of Medical Sciences, Department of Pediatrics and Neonatology, Nagoya 467-8601, Japan g Tokyo Women's Medical University, Institute of Medical Genetics, Tokyo 162-0054, Japan h Tokyo Women's Medical University, Department of Pediatrics, Tokyo 162-8111, Japan i University of Dresden, Children's Hospital, Department of Neuropediatrics, 01307 Dresden, Germany -

Beta-Aminoisobutyric Acid As a Novel Regulator of Carbohydrate and Lipid Metabolism
nutrients Review Beta-Aminoisobutyric Acid as a Novel Regulator of Carbohydrate and Lipid Metabolism Dmitrii A. Tanianskii 1,2 , Natalia Jarzebska 3, Andreas L. Birkenfeld 4, John F. O’Sullivan 4,5 and Roman N. Rodionov 3,* 1 Department of Biochemistry, Institute of Experimental Medicine, Acad. Pavlov St., 12, 197376 St. Petersburg, Russia; [email protected] 2 Department of Fundamental Medicine and Medical Technology, St.Petersburg State University, 8 liter A, 21st Line V.O., 199034 St. Petersburg, Russia 3 University Center for Vascular Medicine, Technische Universität Dresden, Fetscherstraße 74, 01307 Dresden, Germany; [email protected] 4 Medical Clinic III, Technische Universität Dresden, Fetscherstraße 74, 01307 Dresden, Germany; [email protected] (A.L.B.); [email protected] (J.F.O.) 5 Charles Perkins Centre and Heart Research Institute, The University of Sydney, 7 Eliza St, Newtown NSW, Sydney 2042, Australia * Correspondence: [email protected]; Tel.: +49-351-458-3659 Received: 5 February 2019; Accepted: 26 February 2019; Published: 28 February 2019 Abstract: The prevalence and incidence of metabolic syndrome is reaching pandemic proportions worldwide, thus warranting an intensive search for novel preventive and treatment strategies. Recent studies have identified a number of soluble factors secreted by adipocytes and myocytes (adipo-/myokines), which link sedentary life style, abdominal obesity, and impairments in carbohydrate and lipid metabolism. In this review, we discuss the metabolic roles of the recently discovered myokine β-aminoisobutyric acid (BAIBA), which is produced by skeletal muscle during physical activity. In addition to physical activity, the circulating levels of BAIBA are controlled by the mitochondrial enzyme alanine: glyoxylate aminotransferase 2 (AGXT2), which is primarily expressed in the liver and kidneys.