ß-Ureidopropionase Deficiency
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Proteomic Approach to Uncover Neuroprotective Mechanisms of Oleocanthal Against Oxidative Stress
International Journal of Molecular Sciences Article A Proteomic Approach to Uncover Neuroprotective Mechanisms of Oleocanthal against Oxidative Stress Laura Giusti 1,†, Cristina Angeloni 2,†, Maria Cristina Barbalace 3, Serena Lacerenza 4, Federica Ciregia 5, Maurizio Ronci 6 ID , Andrea Urbani 7, Clementina Manera 4, Maria Digiacomo 4 ID , Marco Macchia 4, Maria Rosa Mazzoni 4, Antonio Lucacchini 1 ID and Silvana Hrelia 3,* ID 1 Department of Clinical and Experimental Medicine, University of Pisa, 56126 Pisa, Italy; [email protected] (L.G.); [email protected] (A.L.) 2 School of Pharmacy, University of Camerino, 62032 Camerino, Italy; [email protected] 3 Department for Life Quality Studies, Alma Mater Studiorum, University of Bologna, 47921 Rimini, Italy; [email protected] 4 Department of Pharmacy, University of Pisa, 56126 Pisa, Italy; [email protected] (S.L.); [email protected] (C.M.); [email protected] (M.D.); [email protected] (M.M.); [email protected] (M.R.M.) 5 Department of Rheumatology, GIGA Research, Centre Hospitalier Universitaire (CHU) de Liège, University of Liège, 4000 Liège, Belgium; [email protected] 6 Department of Medical, Oral and Biotechnological Sciences, University G. d’Annunzio of Chieti-Pescara, 65127 Pescara, Italy; [email protected] 7 Institute of Biochemistry and Clinical Biochemistry, Catholic University, 00198 Rome, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-209-1235 † These authors contributed equally to this work. Received: 3 July 2018; Accepted: 1 August 2018; Published: 8 August 2018 Abstract: Neurodegenerative diseases represent a heterogeneous group of disorders that share common features like abnormal protein aggregation, perturbed Ca2+ homeostasis, excitotoxicity, impairment of mitochondrial functions, apoptosis, inflammation, and oxidative stress. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Dihydropyrimidinase Deficiency
Dihydropyrimidinase deficiency Description Dihydropyrimidinase deficiency is a disorder that can cause neurological and gastrointestinal problems in some affected individuals. Other people with dihydropyrimidinase deficiency have no signs or symptoms related to the disorder, and in these individuals the condition can be diagnosed only by laboratory testing. The neurological abnormalities that occur most often in people with dihydropyrimidinase deficiency are intellectual disability, seizures, and weak muscle tone (hypotonia). An abnormally small head size (microcephaly) and autistic behaviors that affect communication and social interaction also occur in some individuals with this condition. Gastrointestinal problems that occur in dihydropyrimidinase deficiency include backflow of acidic stomach contents into the esophagus (gastroesophageal reflux) and recurrent episodes of vomiting (cyclic vomiting). Affected individuals can also have deterioration ( atrophy) of the small, finger-like projections (villi) that line the small intestine and provide a large surface area with which to absorb nutrients. This condition, called villous atrophy, can lead to difficulty absorbing nutrients from foods (malabsorption), resulting in a failure to grow and gain weight at the expected rate (failure to thrive). People with dihydropyrimidinase deficiency, including those who otherwise exhibit no symptoms, may be vulnerable to severe, potentially life-threatening toxic reactions to certain drugs called fluoropyrimidines that are used to treat cancer. Common examples of these drugs are 5-fluorouracil and capecitabine. These drugs may not be broken down efficiently and can build up to toxic levels in the body (fluoropyrimidine toxicity), leading to drug reactions including gastrointestinal problems, blood abnormalities, and other signs and symptoms. Frequency Dihydropyrimidinase deficiency is thought to be a rare disorder. -

Dihydropyrimidine Dehydrogenase in the Metabolism of the Anticancer Drugs
Cancer Chemotherapy and Pharmacology (2019) 84:1157–1166 https://doi.org/10.1007/s00280-019-03936-w REVIEW ARTICLE Dihydropyrimidine dehydrogenase in the metabolism of the anticancer drugs Vinay Sharma1 · Sonu Kumar Gupta1 · Malkhey Verma1 Received: 3 May 2019 / Accepted: 21 August 2019 / Published online: 4 September 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Cancer caused by fundamental defects in cell cycle regulation leads to uncontrolled growth of cells. In spite of the treatment with chemotherapeutic agents of varying nature, multiple resistance mechanisms are identifed in cancer cells. Similarly, numerous variations, which decrease the metabolism of chemotherapeutics agents and thereby increasing the toxicity of anticancer drugs have been identifed. 5-Fluorouracil (5-FU) is an anticancer drug widely used to treat many cancers in the human body. Its broad targeting range is based upon its capacity to act as a uracil analogue, thereby disrupting RNA and DNA synthesis. Dihydropyrimidine dehydrogenase (DPD) is an enzyme majorly involved in the metabolism of pyrimidines in the human body and has the same metabolising efect on 5-FU, a pyrimidine analogue. Multiple mutations in the DPD gene have been linked to 5-FU toxicity and inadequate dosages. DPD inhibitors have also been used to inhibit excessive degradation of 5-FU for meeting appropriate dosage requirements. This article focusses on the role of dihydropyrimidine dehydrogenase in the metabolism of the anticancer drug 5-FU and other associated drugs. Keywords Cancer · Anticancer drugs · Dihydropyrimidine dehydrogenase (DPD) · 5-Fluorouracil (5-FU) · Drug resistance · Drug metabolism Introduction by Dihydropyrimidine dehydrogenase (DPD) through the pyrimidine degradation pathway. -
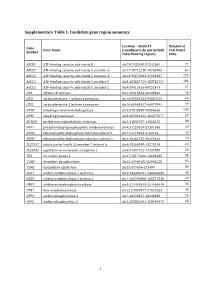
Supplementary Table 1. Candidate Gene Region Summary
Supplementary Table 1. Candidate gene region summary Location – Build 37 Number of Gene Gene Name (coordinates do not include Test Panel Symbol 25kb flanking region) SNPs ABCB1 ATP-binding cassette, sub-family B chr7:87132948-87342564 77 ABCC3 ATP-binding cassette, sub-family C, member 3 chr17:48712218-48769063 64 ABCC4 ATP-binding cassette, sub-family C, member 4 chr13:95672083-95953687 224 ABCC5 ATP-binding cassette, sub-family C, member 5 chr3:183637724-183735727 101 ABCG2 ATP-binding cassette, sub-family G, member 2 chr4:89011416-89152474 57 CDA cytidine deaminase chr1:20915444-20945400 26 CES1 carboxylesterase 1 isoform a precursor chr16:55836764-55867075 24 CES2 carboxylesterase 2 isoform a precursor chr16:66968347-66978994 59 DPYD dihydropyrimidine dehydrogenase chr1:97543300-98386615 239 DPYS dihydropyrimidinase chr8:105391652-105479277 69 MTHFR methylenetetrahydrofolate reductase chr1:11845787-11866115 38 PPAT phosphoribosyl pyrophosphate amidotransferase chr4:57259529-57301845 29 RRM1 ribonucleoside-diphosphate reductase subunit 1 chr11:4115924-4160106 29 RRM2 ribonucleoside-diphosphate reductase subunit 2 chr2:10262735-10270623 19 SLC22A7 solute carrier family 22 member 7 isoform b chr6:43265998-43273276 26 SLC29A1 equilibrative nucleoside transporter 1 chr6:44187242-44201888 26 TK1 thymidine kinase 1 chr17:76170160-76183285 35 TYMP thymidine phosphorylase chr22:50964182-50968258 92 TYMS thymidylate synthetase chr18:657604-673499 34 UCK1 uridine-cytidine kinase 1 isoform a chr9:134399191-134406655 43 UCK2 uridine-cytidine kinase 2 isoform a chr1:165796890-165877339 22 UMPS uridine monophosphate synthase chr3:124449213-124464040 34 UPB1 beta-ureidopropionase chr22:24890077-24922553 30 UPP1 uridine phosphorylase 1 chr7:48128355-48148330 16 UPP2 uridine phosphorylase 2 chr2:158851691-158992478 43 1 Supplementary Table 2. -

Genetic Factors Influencing Pyrimidine- Antagonist Chemotherapy
The Pharmacogenomics Journal (2005) 5, 226–243 & 2005 Nature Publishing Group All rights reserved 1470-269X/05 $30.00 www.nature.com/tpj REVIEW Genetic factors influencing Pyrimidine- antagonist chemotherapy JG Maring1 ABSTRACT 2 Pyrimidine antagonists, for example, 5-fluorouracil (5-FU), cytarabine (ara-C) HJM Groen and gemcitabine (dFdC), are widely used in chemotherapy regimes for 2 FM Wachters colorectal, breast, head and neck, non-small-cell lung cancer, pancreatic DRA Uges3 cancer and leukaemias. Extensive metabolism is a prerequisite for conversion EGE de Vries4 of these pyrimidine prodrugs into active compounds. Interindividual variation in the activity of metabolising enzymes can affect the extent of 1Department of Pharmacy, Diaconessen Hospital prodrug activation and, as a result, act on the efficacy of chemotherapy Meppel & Bethesda Hospital Hoogeveen, Meppel, treatment. Genetic factors at least partly explain interindividual variation in 2 The Netherlands; Department of Pulmonary antitumour efficacy and toxicity of pyrimidine antagonists. In this review, Diseases, University of Groningen & University Medical Center Groningen, Groningen, The proteins relevant for the efficacy and toxicity of pyrimidine antagonists will Netherlands; 3Department of Pharmacy, be summarised. In addition, the role of germline polymorphisms, tumour- University of Groningen & University Medical specific somatic mutations and protein expression levels in the metabolic Center Groningen, Groningen, The Netherlands; pathways and clinical pharmacology -

Genome-Wide Transcriptional Analysis of Carboplatin Response in Chemosensitive and Chemoresistant Ovarian Cancer Cells
1605 Genome-wide transcriptional analysis of carboplatin response in chemosensitive and chemoresistant ovarian cancer cells David Peters,1,2 John Freund,2 and Robert L. Ochs2 variety of disease processes, including cancer. Patterns of global gene expression can reveal the molecular pathways 1 Department of Pharmacology and Therapeutics, University of relevant to the disease process and identify potential new Liverpool, United Kingdom and 2Precision Therapeutics, Inc., Pittsburgh, Pennsylvania therapeutic targets. The use of this technology for the molecular classification of cancer was recently shown with the identification of an expression profile that was Abstract predictive of patient outcome for B-cell lymphoma (1). We have recently described an ex vivo chemoresponse In addition, this study showed that histologically similar assay for determining chemosensitivity in primary cultures tumors can be differentiated based on their gene expression of human tumors. In this study, we have extended these profiles. Ultimately, these unique patterns of gene expres- experiments in an effort to correlate chemoresponse data sion may be used as guidelines to direct different modes of with gene expression patterns at the level of transcription. therapy. Primary cultures of cells derived from ovarian carcinomas Although it is widely recognized that patients with the of individual patients (n = 6) were characterized using same histologic stage and grade of cancer respond to the ChemoFx assay and classified as either carboplatin therapies differently, few clinical tests can predict indi- sensitive (n = 3) or resistant (n = 3). Three representa- vidual patient responses. The next great challenge will be tive cultures of cells from each individual tumor were then to use the power of post-genomic technology, including subjected to Affymetrix gene chip analysis (n = 18) using microarray analyses, to correlate gene expression patterns U95A human gene chip arrays. -

Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience-Dependent Cortical Plasticity
New Research Development Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience- Dependent Cortical Plasticity Milo R. Smith1,2,3,4,5,6,7,8, Poromendro Burman1,3,4,5,8, Masato Sadahiro1,3,4,5,6,8, Brian A. Kidd,2,7 Joel T. Dudley,2,7 and Hirofumi Morishita1,3,4,5,8 DOI:http://dx.doi.org/10.1523/ENEURO.0240-16.2016 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 2Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 3Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 4Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 5Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 6Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 7Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, and 8Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029 Visual Abstract Throughout childhood and adolescence, periods of heightened neuroplasticity are critical for the development of healthy brain function and behavior. Given the high prevalence of neurodevelopmental disorders, such as autism, identifying disruptors of developmental plasticity represents an essential step for developing strategies for prevention and intervention. Applying a novel computational approach that systematically assessed connections between 436 transcriptional signatures of disease and multiple signatures of neuroplasticity, we identified inflammation as a common pathological process central to a diverse set of diseases predicted to dysregulate Significance Statement During childhood and adolescence, heightened neuroplasticity allows the brain to reorganize and adapt to its environment. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -

Potential Widespread Denitrosylation of Brain Proteins Following Prolonged Restraint: Proposed Links Between Stress and Central Nervous System Disease
Metabolic Brain Disease (2019) 34:183–189 https://doi.org/10.1007/s11011-018-0340-1 ORIGINAL ARTICLE Potential widespread denitrosylation of brain proteins following prolonged restraint: proposed links between stress and central nervous system disease Timothy D. Foley1 & Kari S. Koval1 & Alexandria G. Gallagher1 & Stefan H. Olsen1 Received: 9 September 2018 /Accepted: 4 November 2018 /Published online: 9 November 2018 # Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract The biochemical pathways by which aberrant psychophysiological stress promotes neuronal damage and increases the risks for central nervous system diseases are not well understood. In light of previous findings that psychophysiological stress, modeled by animal restraint, can increase the activities and expression levels of nitric oxide synthase isoforms in multiple brain regions, we examined the effects of restraint, for up to 6 h, on levels of S-nitrosylated proteins and NOx (nitrite + nitrate), a marker for high- level nitric oxide generation, in the brains of rats. Results identify functionally-diverse protein targets of S-nitrosylation in the brain, in vivo, and demonstrate the potential for widespread loss of protein nitrosothiols following prolonged restraint despite a concomitant increase in NOx levels. Since physiological levels of protein S-nitrosylation can protect neurons by maintaining redox homeostasis, by limiting excitatory neurotransmission, and by inhibiting apoptotic and inflammatory pathways, we propose that over-activation of protein denitrosylation pathways following sustained or repeated stress may facilitate neural damage and early stages of stress-related central nervous system disease. Keywords Denitrosylation . S-Nitrosylation . Nitric oxide . Psychophysiological stress Introduction the resilience and sensitivity of the CNS to stress remain to be fully established. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2.