The Integrated Relationships of Protriptyline, Thyroid Status and Noradrenergic Mechanisms in Rat Brain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MDPV Bath Salts Test
One Step Methylenedioxypyrovalerone Drug of Abuse Test MATERIALS Analytical Sensitivity The cut-off concentration of the One Step Methylenedioxypyrovalerone Drug of Abuse Test is determined to be 1,000ng/mL. (Dip Card) Materials Provided: Test was run in 30 replicates with negative urine and standard control at ±25% cut-off and ±50% cut-off concentration levels. Test results ● Dip cards are summarized below. For Forensic Use Only ● Desiccants ● Package insert Test Result INTENDED USE Percent of Cut-off n Materials Required But Not Provided: Methylenedioxypyrovalerone Concentration in ng/mL The One Step Methylenedioxypyrovalerone Drug of Abuse Test is a lateral flow chromatographic immunoassay for the ● Specimen collection container Negative Positive qualitative detection of Methylenedioxypyrovalerone (MDPV) in human urine specimen at the cut-off level of 1,000ng/mL. This ● Disposable gloves 0% Cut-off assay is intended for forensic use only. ● Timer 30 30 0 This assay provides only a preliminary qualitative test result. A more specific confirmatory reference method, such as Liquid (No Drug Present) chromatogra -50% Cut-off phy tandem mass spectrometry (LC/MS/MS) or gas chromatography/mass spectrometry (GC/MS) must be use in INSTRUCTIONS FOR USE] 30 30 0 order to obtain a confirmed analytical result. (500ng/mL) 1) Remove the dip card from the foil pouch. -25% Cut-off 30 30 0 BACKGROUND 2) Remove the cap from the dip card. Label the device with patient or control identifications. (750ng/mL) 3) Immerse the absorbent tip into the urine sample for 5 seconds. Urine sample should not touch the plastic device. Cut-off ‘Bath salts’, a form of designer drugs, also promoted as ‘plant food’ or ‘research chemicals’, is sold mainly in head shops, on 4) Replace the cap over the absorbent tip and lay the dip card on a clean, flat, and non-absorptive surface. -

Octopamine and Tyramine Regulate the Activity of Reproductive Visceral Muscles in the Adult Female Blood-Feeding Bug, Rhodnius Prolixus Sam Hana* and Angela B
© 2017. Published by The Company of Biologists Ltd | Journal of Experimental Biology (2017) 220, 1830-1836 doi:10.1242/jeb.156307 RESEARCH ARTICLE Octopamine and tyramine regulate the activity of reproductive visceral muscles in the adult female blood-feeding bug, Rhodnius prolixus Sam Hana* and Angela B. Lange ABSTRACT Monastirioti et al., 1996). Octopamine and tyramine signal via The role of octopamine and tyramine in regulating spontaneous G-protein coupled receptors (GPCRs), leading to changes in second contractions of reproductive tissues was examined in the messenger levels. The recently updated receptor classification α β female Rhodnius prolixus. Octopamine decreased the amplitude of (Farooqui, 2012) divides the receptors into Oct -R, Oct -Rs β β β spontaneous contractions of the oviducts and reduced RhoprFIRFa- (Oct 1-R, Oct 2-R, Oct 3-R), TYR1-R and TYR2-R. In general, β α induced contractions in a dose-dependent manner, whereas tyramine Oct -Rs lead to elevation of cAMP while Oct -R and TYR-Rs lead to 2+ only reduced the RhoprFIRFa-induced contractions. Both octopamine an increase in Ca (Farooqui, 2012). and tyramine decreased the frequency of spontaneous bursal The movement of eggs in the reproductive system of contractions and completely abolished the contractions at Rhodnius prolixus starts at the ovaries, the site of egg maturation. 5×10−7 mol l−1 and above. Phentolamine, an octopamine receptor Upon ovulation, mature eggs are released into the oviducts antagonist, attenuated the inhibition induced by octopamine on the (Wigglesworth, 1942). Eggs are then guided, via oviductal oviducts and the bursa. Octopamine also increased the levels of peristaltic and phasic contractions, to the common oviduct, where cAMP in the oviducts, and this effect was blocked by phentolamine. -
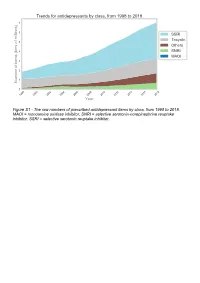
The Raw Numbers of Prescribed Antidepressant Items by Class, from 1998 to 2018
Figure S1 - The raw numbers of prescribed antidepressant items by class, from 1998 to 2018. MAOI = monoamine oxidase inhibitor, SNRI = selective serotonin-norepinephrine reuptake inhibitor, SSRI = selective serotonin reuptake inhibitor. Figure S2 - The raw numbers of prescribed antidepressant items for the ten most commonly prescribed antidepressants (in 2018), from 1998 to 2018. Figure S3 - Practice level percentile charts for the proportions of the ten most commonly prescribed antidepressants (in 2018), prescribed between August 2010 and November 2019. Deciles are in blue, with median shown as a heavy blue line, and extreme percentiles are in red. Figure S4 - Practice level percentile charts for the proportions of specific MAOIs prescribed between August 2010 and November 2019. Deciles are in blue, with median shown as a heavy blue line, and extreme percentiles are in red. Figure S5 - CCG level heat map for the percentage proportions of MAOIs prescribed between December 2018 and November 2019. MAOI = monoamine oxidase inhibitor. Figure S6 - CCG level heat maps for the percentage proportions of paroxetine, dosulepin, and trimipramine prescribed between December 2018 and November 2019. Table S1 - Antidepressant drugs, by category. MAOI = monoamine oxidase inhibitor, SNRI = selective serotonin-norepinephrine reuptake inhibitor, SSRI = selective serotonin reuptake inhibitor. Class Drug MAOI Isocarboxazid, moclobemide, phenelzine, tranylcypromine SNRI Duloxetine, venlafaxine SSRI Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, -

Can Hepatic Coma Be Caused by a Reduction of Brain Noradrenaline Or Dopamine?
Gut: first published as 10.1136/gut.18.9.688 on 1 September 1977. Downloaded from Gut, 1977, 18, 688-691 Can hepatic coma be caused by a reduction of brain noradrenaline or dopamine? L. ZIEVE AND R. L. OLSEN From the Department ofMedicine, Minneapolis Veterans Hospital, University of Minnesota, Minneapolis, and Department of Chemistry, Hamline University, St. Paul, Minnesota, USA SUMMARY Intraventricular infusions of octopamine which raised brain octopamine concentrations more than 20 000-fold resulted in reductions in brain noradrenaline and dopamine by as much as 90% without affecting the alertness or activity of normal rats. As this reduction of brain catechol- amines is much greater than any reported in hepatic coma, we do not believe that values observed in experimental hepatic failure have aetiological significance for the encephalopathy that ensues. Though catecholaminergic nerve terminals represent dopamine and noradrenaline had no discernible only a small proportion of brain synapses (Snyder et effect on the state of alertness of the animals. al., 1973), the reduction in brain dopamine or nor- adrenaline by the accumulation of false neuro- Methods transmitterssuch as octopamine or of aromaticamino acids such as phenylalanine or tyrosine has been ANIMALS suggested as a cause of hepatic coma (Fischer and Male Sprague-Dawley rats weighing between 300 and Baldessarini, 1971; Dodsworth et al., 1974; Fischer 350 were g prepared by the method of Peterson and http://gut.bmj.com/ and Baldessarini, 1975; Munro et al., 1975). The Sparber (1974) for intraventricular infusions of following data have been cited in support of this octopamine. Rats of 250-400 g were used as controls. -

Octopamine Regulates Sleep Indrosophilathrough Protein Kinase A-Dependent Mechanisms
The Journal of Neuroscience, September 17, 2008 • 28(38):9377–9385 • 9377 Behavioral/Systems/Cognitive Octopamine Regulates Sleep in Drosophila through Protein Kinase A-Dependent Mechanisms Amanda Crocker and Amita Sehgal Department of Neuroscience, Howard Hughes Medical Institute, University of Pennsylvania, Philadelphia, Pennsylvania 19104 Sleep is a fundamental process, but its regulation and function are still not well understood. The Drosophila model for sleep provides a powerful system to address the genetic and molecular mechanisms underlying sleep and wakefulness. Here we show that a Drosophila biogenic amine, octopamine, is a potent wake-promoting signal. Mutations in the octopamine biosynthesis pathway produced a pheno- type of increased sleep, which was restored to wild-type levels by pharmacological treatment with octopamine. Moreover, electrical silencing of octopamine-producing cells decreased wakefulness, whereas excitation of these neurons promoted wakefulness. Because protein kinase A (PKA) is a putative target of octopamine signaling and is also implicated in Drosophila sleep, we investigated its role in the effects of octopamine on sleep. We found that decreased PKA activity in neurons rendered flies insensitive to the wake-promoting effects of octopamine. However, this effect of PKA was not exerted in the mushroom bodies, a site previously associated with PKA action on sleep. These studies identify a novel pathway that regulates sleep in Drosophila. Key words: Drosophila; octopamine; sleep; locomotion; norepinephrine; biogenic amine; arousal Introduction The only arousal-promoting signal identified in Drosophila is Sleep is a core process that spans genetically diverse eukaryotes dopamine (Andretic et al., 2005; Kume et al., 2005). from mammals to arthropods (Tobler, 2005). -

Fatal Toxicity of Antidepressant Drugs in Overdose
BRITISH MEDICAL JOURNAL VOLUME 295 24 OCTOBER 1987 1021 Br Med J (Clin Res Ed): first published as 10.1136/bmj.295.6605.1021 on 24 October 1987. Downloaded from PAPERS AND SHORT REPORTS Fatal toxicity of antidepressant drugs in overdose SIMON CASSIDY, JOHN HENRY Abstract dangerous in overdose, thus meriting investigation of their toxic properties and closer consideration of the circumstances in which A fatal toxicity index (deaths per million National Health Service they are prescribed. Recommendations may thus be made that prescriptions) was calculated for antidepressant drugs on sale might reduce the number offatalities. during the years 1975-84 in England, Wales, and Scotland. The We used national mortality statistics and prescription data tricyclic drugs introduced before 1970 had a higher index than the to compile fatal toxicity indices for the currently available anti- mean for all the drugs studied (p<0-001). In this group the depressant drugs to assess the comparative safety of the different toxicity ofamitriptyline, dibenzepin, desipramine, and dothiepin antidepressant drugs from an epidemiological standpoint. Owing to was significantly higher, while that ofclomipramine, imipramine, the nature of the disease these drugs are particularly likely to be iprindole, protriptyline, and trimipramine was lower. The mono- taken in overdose and often cause death. amine oxidase inhibitors had intermediate toxicity, and the antidepressants introduced since 1973, considered as a group, had significantly lower toxicity than the mean (p<0-001). Ofthese newer drugs, maprotiline had a fatal toxicity index similar to that Sources ofinformation and methods of the older tricyclic antidepressants, while the other newly The statistical sources used list drugs under their generic and proprietary http://www.bmj.com/ introduced drugs had lower toxicity indices, with those for names. -

Recent Trends in the Quantification of Biogenic Amines in Biofluids
Journal of Clinical Medicine Review Recent Trends in the Quantification of Biogenic Amines in Biofluids as Biomarkers of Various Disorders: A Review Alina Plenis 1,* , Ilona Ol˛edzka 1 , Piotr Kowalski 1 , Natalia Mi˛ekus 1,2 and Tomasz B ˛aczek 1 1 Department of Pharmaceutical Chemistry, Medical University of Gda´nsk,Hallera 107, 80-416 Gda´nsk, Poland; [email protected] (I.O.); [email protected] (P.K.); [email protected] (N.M.); [email protected] (T.B.) 2 Department of Animal and Human Physiology, Faculty of Biology, University of Gda´nsk,Wita Stwosza 59, 80-308 Gda´nsk,Poland * Correspondence: [email protected]; Fax: +48-58-349-16-35 Received: 4 April 2019; Accepted: 6 May 2019; Published: 9 May 2019 Abstract: Biogenic amines (BAs) are bioactive endogenous compounds which play a significant physiological role in many cell processes like cell proliferation and differentiation, signal transduction and membrane stability. Likewise, they are important in the regulation of body temperature, the increase/decrease of blood pressure or intake of nutrition, as well as in the synthesis of nucleic acids and proteins, hormones and alkaloids. Additionally, it was confirmed that these compounds can be considered as useful biomarkers for the diagnosis, therapy and prognosis of several neuroendocrine and cardiovascular disorders, including neuroendocrine tumours (NET), schizophrenia and Parkinson’s Disease. Due to the fact that BAs are chemically unstable, light-sensitive and possess a high tendency for spontaneous oxidation and decomposition at high pH values, their determination is a real challenge. Moreover, their concentrations in biological matrices are extremely low. -

Association of Selective Serotonin Reuptake Inhibitors with the Risk for Spontaneous Intracranial Hemorrhage
Supplementary Online Content Renoux C, Vahey S, Dell’Aniello S, Boivin J-F. Association of selective serotonin reuptake inhibitors with the risk for spontaneous intracranial hemorrhage. JAMA Neurol. Published online December 5, 2016. doi:10.1001/jamaneurol.2016.4529 eMethods 1. List of Antidepressants for Cohort Entry eMethods 2. List of Antidepressants According to the Degree of Serotonin Reuptake Inhibition eMethods 3. Potential Confounding Variables Included in Multivariate Models eMethods 4. Sensitivity Analyses eFigure. Flowchart of Incident Antidepressant (AD) Cohort Definition and Case- Control Selection eTable 1. Crude and Adjusted Rate Ratios of Intracerebral Hemorrhage Associated With Current Use of SSRIs Relative to TCAs eTable 2. Crude and Adjusted Rate Ratios of Subarachnoid Hemorrhage Associated With Current Use of SSRIs Relative to TCAs eTable 3. Crude and Adjusted Rate Ratios of Intracranial Extracerebral Hemorrhage Associated With Current Use of SSRIs Relative to TCAs. eTable 4. Crude and Adjusted Rate Ratios of Intracerebral Hemorrhage Associated With Current Use of Antidepressants With Strong Degree of Inhibition of Serotonin Reuptake Relative to Weak eTable 5. Crude and Adjusted Rate Ratios of Subarachnoid Hemorrhage Associated With Current Use of Antidepressants With Strong Degree of Inhibition of Serotonin Reuptake Relative to Weak eTable 6. Crude and Adjusted Rate Ratios of Intracranial Extracerebral Hemorrhage Associated With Current Use of Antidepressants With Strong Degree of Inhibition of Serotonin Reuptake Relative to Weak This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/02/2021 eMethods 1. -

Catecholamines and the Hydroxylation of Tyrosine in Synaptosomes Isolated from Rat Brain (DOPA/Tyramine/Dopamine/Norepinephrine/Octopamine) M
Proc. Nat. Acad. Sci. USA Vol. 68, No. 10, pp. 2370-2373, October 1971 Catecholamines and the Hydroxylation of Tyrosine in Synaptosomes Isolated from Rat Brain (DOPA/tyramine/dopamine/norepinephrine/octopamine) M. KAROBATH Psychiatric Research Laboratories, Massachusetts General Hospital, Boston, Mass. 02114 Communicated by Seymour S. Kety, July 16, 1971 ABSTRACT Tyrosine hydroxylase activity of synapto- removing transmitters from an extraneuronal location at the somes isolated from rat brain was examined. A modified synapse, then incubation of synaptosomes with catechol- tritium-displacement assay was used, which allowed the measurement of tyrosine hydroxylase activity without the amines should inhibit the formation of DOPA from tyrosine. addition of either inhibitors of the metabolism of the The experiments demonstrate that tyrosine hydroxylase hydroxylated products or added exogenous cofactor. The activity is affected by catecholamine uptake. The concentra- enzyme activity was strongly inhibited by the addition of tions required to inhibit the synthesis of catechols are in the exogenous catecholamines and 3,4-dihydroxy-L-phenyl- M. alanine. Aromatic amines other than catechols did not range of 10-7 markedly influence tyrosine hydroxylase activity. These MATERIALS AND METHODS in vitro findings support the hypothesis that synthesis of catecholamines is regulated by a mechanism of end- [3,5-H]HifTyrosine (Tracerlab) was purified by column chro- product inhibition at the tyrosine hydroxylase step. matography. Catechol impurities were absorbed on alumina and tritiated water and anions were removed by Synaptosomes are pinched-off nerve endings with relatively columns (8), by non-neuronal elements (1, 2). They absorption on Dowex-50 resin. Tyrosine was eluted from little contamination Dowex-50 with 25 ml of 4 N HCl; after distillation of the eluate contain the enzymes necessary for the synthesis of dopamine (v/v) from tyrosine (3, 4). -

(LSD) Test Dip Card (Urine) • Specimen Collection Container % Agreement 98.8% 99
frozen and stored below -20°C. Frozen specimens should be thawed and mixed before testing. GC/MS. The following results were tabulated: Method GC/MS MATERIALS Total Results Results Positive Negative Materials Provided LSD Rapid Positive 79 1 80 LSD • Test device • Desiccants • Package insert • Urine cups Test Dip card Negative 1 99 100 Materials Required But Not Provided Total Results 80 100 180 One Step Lysergic acid diethylamide (LSD) Test Dip card (Urine) • Specimen collection container % Agreement 98.8% 99. % 98.9% • Timer Package Insert DIRECTIONS FOR USE Analytical Sensitivity This Instruction Sheet is for testing of Lysergic acid diethylamide. Allow the test device, and urine specimen to come to room temperature [15-30°C (59-86°F)] prior to testing. A drug-free urine pool was spiked with LSD at the following concentrations: 0 ng/mL, -50%cutoff, -25%cutoff, cutoff, A rapid, one step test for the qualitative detection of Lysergic acid diethylamide and its metabolites in human urine. 1) Remove the test device from the foil pouch. +25%cutoff and +50%cutoff. The result demonstrates >99% accuracy at 50% above and 50% below the cut-off For forensic use only. 2) Remove the cap from the test device. Label the device with patient or control identifications. concentration. The data are summarized below: INTENDED USE 3) Immerse the absorbent tip into the urine sample for 10-15 seconds. Urine sample should not touch the plastic Lysergic acid diethylamide (LSD) Percent of Visual Result The One Step Lysergic acid diethylamide (LSD) Test Dip card (Urine) is a lateral flow chromatographic device. -

New Zealand Data Sheet
1 PARNATE tranylcypromine film-coated tablets 10 mg New Zealand Data Sheet 1 PARNATE® (10 MG FILM-COATED TABLETS) PARNATE 10 mg film-coated tablets. 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Parnate 10 mg film-coated tablets: each tablet contains tranylcypromine sulfate equivalent to 10 mg of tranylcypromine. Excipients with known effect: each tablet contains sucrose 6 mg. For the full list of excipients, see section 6.1. 3 PHARMACEUTICAL FORM Parnate 10 mg film-coated tablets contain "geranium rose" coloured, biconvex, film-coated tablets. 4 CLINICAL PARTICULARS 4.1 Therapeutic indications Parnate is indicated for the treatment of symptoms of depressive illness especially where treatment with other types of anti-depressants has failed. It is not recommended for use in mild depressive states resulting from temporary situational difficulties. 4.2 Dose and method of administration Adults Begin with 20 mg a day given as 10 mg in the morning and 10 mg in the afternoon. If there is no satisfactory response after two weeks, add one more tablet at midday. Continue this dosage for at least a week. A dosage of 3 tablets a day should only be exceeded with caution. When a satisfactory response is established, dosage may be reduced to a maintenance level. Some patients will be maintained on 20 mg per day, some will need only 10 mg daily. If no improvement occurs, continued administration is unlikely to be beneficial. 1 2 PARNATE tranylcypromine film-coated tablets 10 mg When given together with a tranquilliser, the dosage of Parnate is not affected. When the medicine is given concurrently with electroconvulsive therapy, the recommended dosage is 10 mg twice a day during the series and 10 mg a day afterwards as maintenance therapy. -

Amphetamine, 3,4
0026-895X/01/6006-1181–1188$3.00 MOLECULAR PHARMACOLOGY Vol. 60, No. 6 Copyright © 2001 The American Society for Pharmacology and Experimental Therapeutics 1304/951292 Mol Pharmacol 60:1181–1188, 2001 Printed in U.S.A. ACCELERATED COMMUNICATION Amphetamine, 3,4-Methylenedioxymethamphetamine, Lysergic Acid Diethylamide, and Metabolites of the Catecholamine Neurotransmitters Are Agonists of a Rat Trace Amine Receptor JAMES R. BUNZOW, MARK S. SONDERS, SEKSIRI ARTTAMANGKUL, LAURA M. HARRISON, GE ZHANG, DENISE I. QUIGLEY, TRISTAN DARLAND, KATHERINE L. SUCHLAND, SHAILAJA PASUMAMULA, JAMES L. KENNEDY, SUSAN B. OLSON, R. ELLEN MAGENIS, SUSAN G. AMARA, and DAVID K. GRANDY Departments of Physiology & Pharmacology (J.R.B., S.A., L.M.H., G.Z., D.I.Q., T.D., K.L.S., S.P., S.B.O., R.E.M., D.K.G.) and Molecular and Medical Genetics (S.B.O., R.E.M), School of Medicine, the Vollum Institute (M.S.S., S.G.A.), and the Howard Hughes Medical Institute (S.G.A.), Oregon Health & Science University, Portland, Oregon; and Centre for Addiction and Mental Health, University of Toronto, Canada (J.L.K.) Received August 21, 2001; accepted September 28, 2001 This paper is available online at http://molpharm.aspetjournals.org ABSTRACT The trace amine para-tyramine is structurally and functionally re- goline derivatives, adrenergic ligands, and 3-methylated metabo- lated to the amphetamines and the biogenic amine neurotrans- lites of the catecholamine neurotransmitters are also good ago- mitters. It is currently thought that the biological activities elicited nists at the rat trace amine receptor 1 (rTAR1). These results by trace amines such as p-tyramine and the psychostimulant suggest that the trace amines and catecholamine metabolites amphetamines are manifestations of their ability to inhibit the may serve as the endogenous ligands of a novel intercellular clearance of extracellular transmitter and/or stimulate the efflux of signaling system found widely throughout the vertebrate brain and transmitter from intracellular stores.