A Novel Function for 12-Lipoxygenase in C-Met and Integrin Β4 Axis Crosstalk Elizabeth Tovar Wayne State University
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Integrated Multivariate Analysis of Transcriptomic Data Reveals Immunological Mechanisms in Mice After Leishmania Infantum Infec
1 Integrated multivariate analysis of transcriptomic data 2 reveals immunological mechanisms in mice after 3 Leishmania infantum infection 4 5 Génesis Palacios1, Raquel Diaz-Solano1, Basilio Valladares1,2,4, Roberto Dorta- 6 Guerra1,3, Emma Carmelo1,2,4*. 7 1 Instituto Universitario de Enfermedades Tropicales y Salud Pública de Canarias, 8 Universidad de la Laguna, La Laguna, Islas Canarias, Spain.2 Departamento de 9 Obstetricia y Ginecología, Pediatría, Medicina Preventiva y Salud Pública, 10 Toxicología, Medicina Legal y Forense y Parasitología. 3 Departamento de 11 Matemáticas, Estadística e Investigación Operativa, Facultad de Ciencias, 12 Universidad de La Laguna, La Laguna, Spain. 4 Red de Investigación Colaborativa 13 en Enfermedades Tropicales (RICET). 14 15 16 17 18 19 20 21 1 22 SUPPLEMENTAL MATERIAL 23 Supplementary file 1. List of TaqMan assays used for RT-qPCR analysis using 24 QuantStudioTM 12K Flex Real-Time PCR System. 25 PANEL 1. N° Assay ID GEN NOMBRE DEL GEN GRUPO 1 Mm00437762_m1 B2m beta-2 microglobulin 2 Mm00446968_m1 Hprt hypoxanthine guanine phosphoribosyl transferase 3 Mm00435617_m1 Pgk1 phosphoglycerate kinase 1 Endogenous gene 4 Mm01277042_m1 Tbp TATA box binding protein expression 5 Mm01201237_m1 Ubc ubiquitin C tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation 6 Mm01722325_m1 Ywhaz protein, zeta polypeptide 7 Mm00599890_m1 Ifngr1 interferon gamma receptor 1 8 Mm00492626_m1 Ifngr2 interferon gamma receptor 2 9 Mm01168134_m1 Ifng interferon gamma 10 Mm01178820_m1 Tgfb1 transforming growth factor, -

Table S2.Up Or Down Regulated Genes in Tcof1 Knockdown Neuroblastoma N1E-115 Cells Involved in Differentbiological Process Anal
Table S2.Up or down regulated genes in Tcof1 knockdown neuroblastoma N1E-115 cells involved in differentbiological process analysed by DAVID database Pop Pop Fold Term PValue Genes Bonferroni Benjamini FDR Hits Total Enrichment GO:0044257~cellular protein catabolic 2.77E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 537 13588 1.944851 8.64E-07 8.64E-07 5.02E-07 process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0051603~proteolysis involved in 4.52E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 534 13588 1.93519 1.41E-06 7.04E-07 8.18E-07 cellular protein catabolic process ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, USP17L5, FBXO11, RAD23B, NEDD8, UBE2V2, RFFL, CDC GO:0044265~cellular macromolecule 6.09E-10 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 609 13588 1.859332 1.90E-06 6.32E-07 1.10E-06 catabolic process ISG15, RBM8A, ATG7, LOC100046898, PSENEN, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, ASB8, DCUN1D1, PSMA6, SIAH1A, TRIM32, RNF138, GM12396, RNF20, XRN2, USP17L5, FBXO11, RAD23B, UBE2V2, NED GO:0030163~protein catabolic process 1.81E-09 MKRN1, PPP2R5C, VPRBP, MYLIP, CDC16, ERLEC1, MKRN2, CUL3, 556 13588 1.87839 5.64E-06 1.41E-06 3.27E-06 ISG15, ATG7, PSENEN, LOC100046898, CDCA3, ANAPC1, ANAPC2, ANAPC5, SOCS3, ENC1, SOCS4, -

PTGS1, PTGS2, ALOX5, ALOX12, ALOX15, and FLAP Snps: Interaction with Fatty Acids in Colon Cancer and Rectal Cancer
Genes Nutr (2013) 8:115–126 DOI 10.1007/s12263-012-0302-x RESEARCH PAPER PTGS1, PTGS2, ALOX5, ALOX12, ALOX15, and FLAP SNPs: interaction with fatty acids in colon cancer and rectal cancer Nina Habermann • Cornelia M. Ulrich • Abbie Lundgreen • Karen W. Makar • Elizabeth M. Poole • Bette Caan • Richard Kulmacz • John Whitton • Rachel Galbraith • John D. Potter • Martha L. Slattery Received: 3 February 2012 / Accepted: 18 May 2012 / Published online: 8 June 2012 Ó Springer-Verlag 2012 Abstract Dietary polyunsaturated fatty acids (PUFAs) for low docosahexaenoic acid intake among those with the can be converted to prostaglandins and leukotrienes. PTGS1 rs10306110 (-1,053 A [ G) variant genotypes Oxygenation of omega-6 PUFAs generally results in the (OR = 1.6, 95 % confidence interval = 1.1–2.3, adj. production of pro-inflammatory mediators, whereas oxy- p = 0.06) and rectal cancer risk for low total fat intake genated products of omega-3 (n-3) PUFAs generally have among those with the variant PTGS1 rs10306122 (7,135 lower inflammatory activity. We hypothesize that elevated A [ G) (ORvs.wt = 1.80, 1.02–2.99; adj. p = 0.08). The n-3 PUFA intakes from fish are associated with lower risk ALOX15 rs11568131 (10,339 C [ T) wild type in combi- of colorectal cancer among those with genetic variants that nation with a high inflammation score (low EPA intake, result in higher levels of pro-inflammatory mediators. In high AA intake, no regular NSAID use, high BMI, smok- population-based case–control studies of colon (case ing) was associated with increased colon cancer risk n = 1,574) and rectal cancer (case n = 791) and disease- (OR = 2.28, 1.7–3.07). -

Inhibition of Oxidative Stress and ALOX12 and NF-B Pathways
antioxidants Article Inhibition of Oxidative Stress and ALOX12 and NF-κB Pathways Contribute to the Protective Effect of Baicalein on Carbon Tetrachloride-Induced Acute Liver Injury Chongshan Dai 1,2,* , Hui Li 3, Yang Wang 1,2, Shusheng Tang 1,2, Tony Velkov 4,* and Jianzhong Shen 1,2 1 College of Veterinary Medicine, China Agricultural University, No. 2 Yuanmingyuan West Road, Beijing 100193, China; [email protected] (Y.W.); [email protected] (S.T.); [email protected] (J.S.) 2 Beijing Key Laboratory of Detection Technology for Animal-Derived Food Safety, College of Veterinary Medicine, China Agricultural University, Beijing 100193, China 3 Beijing Key Laboratory of Diagnostic and Traceability Technologies for Food Poisoning, Beijing Center for Disease Prevention and Control, Beijing 100193, China; [email protected] 4 Department of Pharmacology & Therapeutics, School of Biomedical Sciences, Faculty of Medicine, Dentistry and Health Sciences, The University of Melbourne, Parkville, VIC 3010, Australia * Correspondence: [email protected] (C.D.); [email protected] (T.V.) Abstract: This study investigates the protective effect of baicalein on carbon tetrachloride (CCl4)-induced acute liver injury and the underlying molecular mechanisms. Mice were orally administrated baicalein at 25 and 100 mg/kg/day for 7 consecutive days or ferrostatin-1 (Fer-1) at 10 mg/kg was i.p. injected in mice at 2 and 24 h prior to CCl4 injection or the vehicle. Our results showed that baicalein or Fer-1 supplementation significantly attenuated CCl4 exposure-induced elevations of serum alanine Citation: Dai, C.; Li, H.; Wang, Y.; aminotransferase and aspartate aminotransferase, and malondialdehyde levels in the liver tissues and Tang, S.; Velkov, T.; Shen, J. -

Regulation of Tissue Inflammation by 12-Lipoxygenases
biomolecules Review Regulation of Tissue Inflammation by 12-Lipoxygenases Abhishek Kulkarni 1 , Jerry L. Nadler 2, Raghavendra G. Mirmira 1,* and Isabel Casimiro 1,* 1 Department of Medicine, The University of Chicago, Chicago, IL 60637, USA; [email protected] 2 Department of Medicine and Pharmacology, New York Medical College, Valhalla, NY 10595, USA; [email protected] * Correspondence: [email protected] (R.G.M.); [email protected] (I.C.) Abstract: Lipoxygenases (LOXs) are lipid metabolizing enzymes that catalyze the di-oxygenation of polyunsaturated fatty acids to generate active eicosanoid products. 12-lipoxygenases (12-LOXs) primarily oxygenate the 12th carbon of its substrates. Many studies have demonstrated that 12-LOXs and their eicosanoid metabolite 12-hydroxyeicosatetraenoate (12-HETE), have significant pathological implications in inflammatory diseases. Increased level of 12-LOX activity promotes stress (both oxidative and endoplasmic reticulum)-mediated inflammation, leading to damage in these tissues. 12-LOXs are also associated with enhanced cellular migration of immune cells—a characteristic of several metabolic and autoimmune disorders. Genetic depletion or pharmacological inhibition of the enzyme in animal models of various diseases has shown to be protective against disease development and/or progression in animal models in the setting of diabetes, pulmonary, cardiovascular, and metabolic disease, suggesting a translational potential of targeting the enzyme for the treatment of several disorders. In this article, we review the role of 12-LOXs in the pathogenesis of several diseases in which chronic inflammation plays an underlying role. Citation: Kulkarni, A.; Nadler, J.L.; Keywords: 12-lipoxygenases; 12-LOXs; 12/15-lipoxygenase; 12/15-LOX; lipoxygenases; eicosanoids; Mirmira, R.G.; Casimiro, I. -

Original Article Genetic Variants of Aloxs Genes in Polyunsaturated Fatty Acid/Arachidonic Acid Metabolism Associated with Type-2 Diabetes Development
Int J Clin Exp Med 2018;11(12):13797-13805 www.ijcem.com /ISSN:1940-5901/IJCEM0077987 Original Article Genetic variants of ALOXs genes in polyunsaturated fatty acid/arachidonic acid metabolism associated with type-2 diabetes development Jim Jinn-Chyuan Sheu1,3,4,5*, Ying-Ju Lin1,3*, Cherry Yin-Yi Chang2, Shih-Yin Chen1,3, Wen-Ling Liao1,4, Jai-Sing Yang1, Ming-Tsung Lai6, Chih-Mei Chen1, Chun-Cheng Tseng4, Tritium Hwang4, Ping-Ho Chen7, Fuu-Jen Tsai1,3 1Human Genetic Center, 2Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan; 3School of Chinese Medicine, China Medical University, Taichung, Taiwan; 4Institute of Biomedical Sci- ences, National Sun Yat-sen University, Kaohsiung, Taiwan; 5Department of Health and Nutrition Biotechnology, Asia University, Taichung, Taiwan; 6Department of Pathology, Taichung Hospital, Ministry of Health and Welfare, Taichung, Taiwan; 7School of Dentistry, Kaohsiung Medical University, Kaohsiung, Taiwan. *Equal contributors. Received April 16, 2018; Accepted July 24, 2018; Epub December 15, 2018; Published December 30, 2018 Abstract: Poly-unsaturated fatty acids (PUFAs)/arachidonic acids (AAs) and their derived eicosanoids play potent roles in triggering inflammation during obesity and diabetes development. Recent studies have indicated functional roles of ALOX5, ALOX12, ALOX12B, and ALOX15 in the development of insulin resistance and islet β-cell dysfunc- tion. However, the impact of their genetic variants on type 2 diabetes (T2D) development in Asian patients remains unclear. In this study, 1,682 healthy controls and 788 patients with T2D were enrolled for genotyping those four ALOX genes by the TaqMan method. A total of eight Han Chinese-specific SNPs (two SNps for each gene) were selected for this study. -

Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase Is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Spring 2010 Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Michelle Kinder University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Immunology and Infectious Disease Commons Recommended Citation Kinder, Michelle, "Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis" (2010). Publicly Accessible Penn Dissertations. 88. https://repository.upenn.edu/edissertations/88 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/88 For more information, please contact [email protected]. Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Abstract Fatty acid metabolism governs critical cellular processes in multiple cell types. The goal of my dissertation was to investigate the intersection between fatty acid metabolism and hematopoiesis. Although fatty acid metabolism has been extensively studied in mature hematopoietic subsets during inflammation, in developing hematopoietic cells the role of fatty acid metabolism, in particular by 12/ 15-Lipoxygenase (12/15-LOX), was unknown. The observation that 12/15-LOX-deficient (Alox15) mice developed a myeloid leukemia instigated my studies since leukemias are often a consequence of dysregulated hematopoiesis. This observation lead to the central hypothesis of this dissertation which is that polyunsaturated fatty acid metabolism mediated by 12/15-LOX participates in hematopoietic development. Using genetic mouse models and in vitro and in vivo cell development assays, I found that 12/15-LOX indeed regulates multiple stages of hematopoiesis including the function of hematopoietic stem cells (HSC) and the differentiation of B cells, T cells, basophils, granulocytes and monocytes. -

Functional Characterization of Genetic Enzyme Variations in Human Lipoxygenases
Redox Biology 1 (2013) 566–577 Contents lists available at ScienceDirect Redox Biology journal homepage: www.elsevier.com/locate/redox Functional characterization of genetic enzyme variations in human lipoxygenases Thomas Horn a,n, Kumar Reddy Kakularam b, Monika Anton a, Constanze Richter c, Pallu Reddanna b,d, Hartmut Kuhn a a Institute of Biochemistry, University Medicine Berlin—Charité, Charitéplatz 1, D-10117 Berlin, Germany b Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Gachibowli, Hyderabad 500046, Andhra Pradesh, India c Institute of Nutrition Technology and Nutrition Chemistry, TU Berlin, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany d National Institute of Animal Biotechnology, Hyderabad 500046, Andhra Pradesh, India article info abstract Article history: Mammalian lipoxygenases play a role in normal cell development and differentiation but they have also been Received 28 October 2013 implicated in the pathogenesis of cardiovascular, hyperproliferative and neurodegenerative diseases. As lipid Accepted 1 November 2013 peroxidizing enzymes they are involved in the regulation of cellular redox homeostasis since they produce lipid hydroperoxides, which serve as an efficient source for free radicals. There are various epidemiological Keywords: correlation studies relating naturally occurring variationsinthesixhumanlipoxygenasegenes(SNPsorrare Eicosanoids mutations) to the frequency for various diseases in these individuals, but for most of the described variations no Leukotrienes functional data are available. Employing a combined bioinformatical and enzymological strategy, which Lipoxygenases included structural modeling and experimental site-directed mutagenesis, we systematically explored the Gene polymorphism structural and functional consequences of non-synonymous genetic variations in four different human SNP lipoxygenase genes (ALOX5, ALOX12, ALOX15, and ALOX15B) that have been identified in the human 1000 genome project. -

The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2013 The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach Wüst, Sophia Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-92378 Dissertation Published Version Originally published at: Wüst, Sophia. The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Ap- proach. 2013, University of Zurich, Faculty of Medicine. The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr.sc.nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Sophia Julia Annette Wüst von Oberriet-Montlingen SG Promotionskomitee Prof. Dr. Thierry Hennet Prof. Dr. Martin Hersberger (Leitung der Dissertation) Prof. Dr. Arnold von Eckardstein Zürich, 2014 CONTENTS CONTENTS ..............................................................................................1 ABBREVIATIONS.....................................................................................3 SUMMARY................................................................................................6 ZUSAMMENFASSUNG............................................................................8 1. INTRODUCTION ................................................................................10 1.1 Atherosclerosis .................................................................................10 -

And Anti-Inflammatory Metabolites and Its Potential Role in Rheumatoid
cells Review Circulating Pro- and Anti-Inflammatory Metabolites and Its Potential Role in Rheumatoid Arthritis Pathogenesis Roxana Coras 1,2, Jessica D. Murillo-Saich 1 and Monica Guma 1,2,* 1 Department of Medicine, School of Medicine, University of California, San Diego, 9500 Gilman Drive, San Diego, CA 92093, USA; [email protected] (R.C.); [email protected] (J.D.M.-S.) 2 Department of Medicine, Autonomous University of Barcelona, Plaça Cívica, 08193 Bellaterra, Barcelona, Spain * Correspondence: [email protected] Received: 22 January 2020; Accepted: 18 March 2020; Published: 30 March 2020 Abstract: Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease that affects synovial joints, leading to inflammation, joint destruction, loss of function, and disability. Although recent pharmaceutical advances have improved the treatment of RA, patients often inquire about dietary interventions to improve RA symptoms, as they perceive pain and/or swelling after the consumption or avoidance of certain foods. There is evidence that some foods have pro- or anti-inflammatory effects mediated by diet-related metabolites. In addition, recent literature has shown a link between diet-related metabolites and microbiome changes, since the gut microbiome is involved in the metabolism of some dietary ingredients. But diet and the gut microbiome are not the only factors linked to circulating pro- and anti-inflammatory metabolites. Other factors including smoking, associated comorbidities, and therapeutic drugs might also modify the circulating metabolomic profile and play a role in RA pathogenesis. This article summarizes what is known about circulating pro- and anti-inflammatory metabolites in RA. It also emphasizes factors that might be involved in their circulating concentrations and diet-related metabolites with a beneficial effect in RA. -

Cutting Edge
Cutting Edge: Severe SARS-CoV-2 Infection in Humans Is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators Benjamin Schwarz,*,1 Lokesh Sharma,†,1 Lydia Roberts,*,1 Xiaohua Peng,† † ‡ † Santos Bermejo, x Ian Leighton,* Arnau Casanovas-Massana, Maksym Minasyan, Shelli Farhadian, Albert I. Ko,‡ Yale IMPACT Team,2 Charles S. Dela Cruz,†,1 and Catharine M. Bosio*,1 The COVID-19 pandemic has affected more than and converted via enzymatic hydroxylation to immune regulating 20 million people worldwide, with mortality exceeding lipid mediators (LMs) (1–7). LMs function as inflammatory, 800,000 patients. Risk factors associated with severe immune-regulatory, or proresolving immune signals during disease and mortality include advanced age, hyperten- chronic and acute immune responses (1, 8). Previous studies have sion, diabetes, and obesity. Each of these risk factors demonstrated that comorbidities associated with severe COVID- pathologically disrupts the lipidome, including immu- 19 including obesity, hypertension, diabetes and heart disease nomodulatory eicosanoid and docosanoid lipid media- feature pathological disruption of the lipidome including altered tors (LMs). We hypothesized that dysregulation of LMs baseline levels of LMs and the PUFA-containing LM precursors may be a defining feature of the severity of COVID-19. in absence of infection (9–13). Several studies have examined By examining LMs and polyunsaturated fatty acid pre- systemic metabolic correlates of COVID-19, including shifts in cursor lipids in serum from hospitalized COVID-19 pa- the lipidome (14–17) (J. Troisi et al., Research Square, 2020, https://doi.org/10.21203/rs.3.rs-34085/v1). Collectively, these tients, we demonstrate that moderate and severe disease studies demonstrated that severe COVID-19 is marked by a are separated by specific differences in abundance of systemic dysregulation of metabolism and widespread changes in immune-regulatory and proinflammatory LMs. -
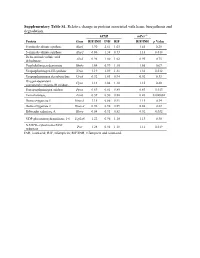
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-