Genome of the Chinese Tree Shrew
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Conservation Behaviorist
The Conservation Behaviorist Heidi Fisher, student at Boston University, receives E. O. Wilson Conservation Award Animal Behavior Society ABS Conservation Committee The Conservation Behaviorist, an electronic biannual news-update, informs ABS members about the Conservation Committee’s activities, research trends in behavior and conservation, and relevant scientific news in conservation research where behavior plays an important role. The Conservation Behaviorist Vol. 3, No. 1, May 2005 ____________________________________________________________ In this issue The ABS Conservation Committee ..……………………….. 2 Encyclopedia of Animal Behavior........................................ 2 Elizabeth V. Lonsdorf and Mark L. Wildhaber join ABS Conservation Committee…………………………………….. 2 Perspectives: Animal Cognition and its Role in Conservation Behavior, by Guillermo Paz-y-Miño C.……………………... 3 Feature Article: Conservation Behavior in Borneo’s logged forests, by Jason Munshi-South….…………………..……….5 Animal Behavior Society Annual Meeting……………………7 Excerpts From Back Issues …………………...…………….10 © Photo Sherri Michaud Conservation Tips By Daniel T. Blumstein “…As a field biologist, it is difficult not to become a conservationist, particularly when Is there anything a behaviorist can you study animal behavior. An animal’s first do to help conservation? response to a stressor is often a change at Work in an endangered habitat. Even if you the behavioral level. Behavior is a reliable are not focusing on an endangered species, indicator of ecological disturbance…” says by working in an endangered habitat you will Fisher. Her proposal “Communication breakdown illustrate, by example, the value of the habitat and hybridization in Xiphophorus fishes” will be and you may be able to collect additional funded by the ABS Student Research Grant information that will be useful for endangered Program and she will receive the 2005 E. -

Positively Selected Genes of the Chinese Tree Shrew (Tupaia Belangeri Chinensis) Locomotion System
Zoological Research 35 (3): 240−248 DOI:10.11813/j.issn.0254-5853.2014.3.240 Positively selected genes of the Chinese tree shrew (Tupaia belangeri chinensis) locomotion system Yu FAN 1, 2, Dan-Dan YU1, Yong-Gang YAO1,2,3,* 1. Key Laboratory of Animal Models and Human Disease Mechanisms of Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology, Kunming, Yunnan 650223, China 2. Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650223, China 3. Kunming Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China Abstract: While the recent release of the Chinese tree shrew (Tupaia belangeri chinensis) genome has made the tree shrew an increasingly viable experimental animal model for biomedical research, further study of the genome may facilitate new insights into the applicability of this model. For example, though the tree shrew has a rapid rate of speed and strong jumping ability, there are limited studies on its locomotion ability. In this study we used the available Chinese tree shrew genome information and compared the evolutionary pattern of 407 locomotion system related orthologs among five mammals (human, rhesus monkey, mouse, rat and dog) and the Chinese tree shrew. Our analyses identified 29 genes with significantly high ω (Ka/Ks ratio) values and 48 amino acid sites in 14 genes showed significant evidence of positive selection in the Chinese tree shrew. Some of these positively selected genes, e.g. HOXA6 (homeobox A6) and AVP (arginine vasopressin), play important roles in muscle contraction or skeletal morphogenesis. These results provide important clues in understanding the genetic bases of locomotor adaptation in the Chinese tree shrew. -

Browsing Genes and Genomes with Ensembl
The Bioinformatics Roadshow Tórshavn, The Faroe Islands 28-29 November 2012 BROWSING GENES AND GENOMES WITH ENSEMBL EXERCISES AND ANSWERS 1 BROWSER 3 BIOMART 8 VARIATION 13 COMPARATIVE GENOMICS 18 2 Note: These exercises are based on Ensembl version 69 (October 2012). After in future a new version has gone live, version 69 will still be available at http://e69.ensembl.org/. If your answer doesn’t correspond with the given answer, please consult the instructor. ______________________________________________________________ BROWSER ______________________________________________________________ Exercise 1 – Exploring a gene (a) Find the human F9 (coagulation factor IX) gene. On which chromosome and which strand of the genome is this gene located? How many transcripts (splice variants) have been annotated for it? (b) What is the longest transcript? How long is the protein it encodes? Has this transcript been annotated automatically (by Ensembl) or manually (by Havana)? How many exons does it have? Are any of the exons completely or partially untranslated? (c) Have a look at the external references for ENST00000218099. What is the function of F9? (d) Is it possible to monitor expression of ENST00000218099 with the ILLUMINA HumanWG_6_V2 microarray? If so, can it also be used to monitor expression of the other two transcripts? (e) In which part (i.e. the N-terminal or C-terminal half) of the protein encoded by ENST00000218099 does its peptidase activity reside? (f) Have any missense variants been discovered for the protein encoded by ENST00000218099? (g) Is there a mouse orthologue predicted for the human F9 gene? (h) If you have yourself a gene of interest, explore what information Ensembl displays about it! ______________________________________________________________ Answer (a) 8 Go to the Ensembl homepage (http://www.ensembl.org/). -

Endemism and Diversity of Small Mammals Along Two Neighboring Bornean Mountains
Endemism and diversity of small mammals along two neighboring Bornean mountains Miguel Camacho-Sanchez1,2,*, Melissa T.R. Hawkins3,4,5,*, Fred Tuh Yit Yu6, Jesus E. Maldonado3 and Jennifer A. Leonard1 1 Conservation and Evolutionary Genetics Group, Doñana Biological Station (EBD-CSIC), Sevilla, Spain 2 CiBIO—Centro de Investigação em Biodiversidade e Recursos Genéticos da Universidade do Porto, Vairão, Portugal 3 Center for Conservation Genomics, Smithsonian Conservation Biology Institute, National Zoological Park, Washington, DC, USA 4 Department of Biological Sciences, Humboldt State University, Arcata, CA, USA 5 Division of Mammals, National Museum of Natural History, Washington, DC, USA 6 Sabah Parks, Kota Kinabalu, Sabah, Malaysia * These authors contributed equally to this work. ABSTRACT Mountains offer replicated units with large biotic and abiotic gradients in a reduced spatial scale. This transforms them into well-suited scenarios to evaluate biogeographic theories. Mountain biogeography is a hot topic of research and many theories have been proposed to describe the changes in biodiversity with elevation. Geometric constraints, which predict the highest diversity to occur in mid-elevations, have been a focal part of this discussion. Despite this, there is no general theory to explain these patterns, probably because of the interaction among different predictors with the local effects of historical factors. We characterize the diversity of small non-volant mammals across the elevational gradient on Mount (Mt.) Kinabalu (4,095 m) and Mt. Tambuyukon (2,579 m), two neighboring mountains in Borneo, Malaysia. We documented a decrease in species richness with elevation which deviates from expectations of the geometric constraints and suggests that spatial Submitted 14 February 2018 Accepted 9 September 2019 factors (e.g., larger diversity in larger areas) are important. -

The Ethmoidal Region of the Skull of Ptilocercus Lowii
Research Article Primate Biol., 2, 89–110, 2015 www.primate-biol.net/2/89/2015/ doi:10.5194/pb-2-89-2015 © Author(s) 2015. CC Attribution 3.0 License. The ethmoidal region of the skull of Ptilocercus lowii (Ptilocercidae, Scandentia, Mammalia) – a contribution to the reconstruction of the cranial morphotype of primates I. Ruf1, S. Janßen2, and U. Zeller2 1Senckenberg Forschungsinstitut und Naturmuseum Frankfurt, Abteilung Paläoanthropologie und Messelforschung, Senckenberganlage 25, 60325 Frankfurt am Main, Germany 2FG Spezielle Zoologie, Lebenswissenschaftliche Fakultät, Albrecht Daniel Thaer-Institut für Agrar- und Gartenbauwissenschaften, Humboldt-Universität zu Berlin, Ziegelstrasse 5–9, 10117 Berlin, Germany Dedicated to Hans-Jürg Kuhn on the occasion of his 80th birthday. Correspondence to: I. Ruf ([email protected]) Received: 17 June 2015 – Revised: 6 September 2015 – Accepted: 7 September 2015 – Published: 25 September 2015 Abstract. The ethmoidal region of the skull houses one of the most important sense organs of mammals, the sense of smell. Investigation of the ontogeny and comparative anatomy of internal nasal structures of the macros- matic order Scandentia is a significant contribution to the understanding of the morphotype of Scandentia with potential implications for our understanding of the primate nasal morphological pattern. For the first time peri- natal and adult stages of Ptilocercus lowii and selected Tupaia species were investigated by serial histological sections and high-resolution computed tomography (µCT), respectively. Scandentia show a very common olfac- tory turbinal pattern of small mammals in having two frontoturbinals, three ethmoturbinals, and one interturbinal between the first and second ethmoturbinal. This indicates a moderately developed sense of smell (moderately macrosmatic). -

Characterization of 12 Polymorphic Microsatellite Markers in the Chinese Tree Shrew (Tupaia Belangeri Chinensis)
Zoological Research 34 (E2): E62−E68 doi: 10.3724/SP.J.1141.2013.E02E62 Characterization of 12 polymorphic microsatellite markers in the Chinese tree shrew (Tupaia belangeri chinensis) Xiao-Hong LIU1, 4, Yong-Gang YAO1, 2, 3,* 1. Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology, Kunming, Yunnan 650223, China; 2. Kunming Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China; 3. Tree Shrew Inbreeding Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China; 4. University of Chinese Academy of Sciences, Beijing 100049, China Abstract: The Chinese tree shrew (Tupaia belangeri chinensis) is a small experimental animal with a close affinity to primates. This species has long been proposed to be an alternative experimental animal to primates in biomedical research. Despite decades of study, there is no pure breed for this animal, and the overall genetic diversity of wild tree shrews remains largely unknown. In order to obtain a set of genetic markers for evaluating the genetic diversity of tree shrew wild populations and tracing the lineages in inbreeding populations, we developed 12 polymorphic microsatellite markers from the genomic DNA of the tree shrew. An analysis of a wild population of 117 individuals collected from the suburb of Kunming, China, showed that these loci exhibited a highly expected heterozygosity (0.616). These 12 microsatellites were sufficient for individual identification and parentage analysis. The microsatellite markers developed in this study will be of use in evaluating genetic diversity and lineage tracing for the tree shrew. -

Creating Animal Models, Why Not Use the Chinese Tree Shrew (Tupaia Belangeri Chinensis)?
ZOOLOGICAL RESEARCH Creating animal models, why not use the Chinese tree shrew (Tupaia belangeri chinensis)? Yong-Gang Yao1,2,* 1 Key Laboratory of Animal Models and Human Disease Mechanisms, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming Yunnan 650223, China 2 Kunming Primate Research Center of the Chinese Academy of Sciences, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming Yunnan 650223, China ABSTRACT the spot light as a viable animal model for investigating the basis of many different human diseases. The Chinese tree shrew (Tupaia belangeri chinensis), a squirrel-like and rat-sized mammal, has a wide Keywords: Chinese tree shrew; Genome biology; distribution in Southeast Asia, South and Southwest Animal model; Gene editing; Innate immunity China and has many unique characteristics that 1 make it suitable for use as an experimental animal. INTRODUCTION There have been many studies using the tree shrew (Tupaia belangeri) aimed at increasing our As human beings, our knowledge about ourselves, especially understanding of fundamental biological mechanisms about how our brain works, how a disease develops, and the and for the modeling of human diseases and discovery of many efficient therapeutic agents, has largely therapeutic responses. The recent release of a come from studies using animals. The higher the similarity publicly available annotated genome sequence of between an animal species and the human, the more we can the Chinese tree shrew and its genome database obtain helpful and precise information concerning the (www.treeshrewdb.org) has offered a solid base fundamental biology, disease mechanism, and safety, efficiency from which it is possible to elucidate the basic and predictability of therapeutic agents (Franco, 2013; biological properties and create animal models using McGonigle & Ruggeri, 2014). -

99Th Annual Meeting & Centennial Celebration Of
99TH ANNUAL MEETING & CENTENNIAL CELEBRATION OF THE AMERICAN SOCIETY OF MAMMALOGISTS 28 JUNE-2 JULY 2019 HYATT REGENCY WASHINGTON ON CAPITOL HILL -WASHINGTON, DC- PROGRAM The 2019 American Society of Mammalogists Annual Meeting logo was designed by John Megahan. It features the African bush elephant (Loxodonta africana), which for decades has welcomed visitors to Smithsonian’s National Museum of Natural History. The Smithsonian Institution Building (aka, The Castle) is featured in the background. 99TH ANNUAL MEETING AND CENTENNIAL CELEBRATION OF THE AMERICAN SOCIETY OF MAMMALOGISTS 28 JUNE – 2 JULY 2019 HYATT REGENCY WASHINGTON ON CAPITOL HILL -WASHINGTON, DC- AMERICAN SOCIETY OF MAMMALOGISTS (ASM) The American Society of Mammalogists (ASM) was established in 1919 for the purpose of promoting interest in the study of mammals. AN OVERVIEW In addition to being among the most charismatic of animals, mammals are important in many disciplines from paleontology to ecology and evolution. We, of course, are mammals and thus are in the interesting position of studying ourselves in quest of a greater understanding of the role of mammals in the natural world. The ASM is currently composed of thousands of members, many of whom are professional scientists. Members of the Society have always had a strong interest in the public good, and this is reflected in their involvement in providing information for public policy, resources management, conservation, and education. The Society hosts annual meetings and maintains several publications. The flagship publication is the Journal of Mammalogy, a journal produced six times per year that accepts submissions on all aspects of mammalogy. The ASM also publishes Mammalian Species (accounts of individual species) and Special Publications (books that pertain to specific taxa or topics), and we maintain a mammal images library that contains many exceptional photographs of mammals. -

Checklist of Mammals of Danum Valley
CHECKLIST OF MAMMALS OF DANUM VALLEY 1. Pangolins MANIDAE - Sunda Pangolin Manis Javanica 2. Mooonrats & allies ERINACIEDAE - Moonrat Echinosorex Gymnurus 3. Tree Shrews TUPAIIDAE - Common Treeshrew Tupaia glis - Lesser Treeshrew Tupaia minor - Striped Treeshrew Tupaia dorsalis - Large Treeshrew Tupaia tana - Painted Treeshrew Tupaia picta - Ruddy Treeshrew Tupaia splendida 4. Flying Lemurs CYNOCEPHALIDAE - Colugo Cynocephalus variegates 5. Fruit-bats PTEROPIDAE - Large Flying Fox Pteropus vampyrus - Island Flying Fox Pteropus hypomelanus 6. Lorises LORIDAE - Slow Loris Nycticebus coucang 7. Tarsiers TARSIIDAE - Western Tarsier Tarsius bancanus 8. Old World Monkeys CERCOPITHECIDAE - Long-tailed Macaque Macaca fascicularis - Pig-tailed Macaque Macaca nemestrina - Proboscis Monkey Nasalis larvatus - Red Leaf Monkey Presbytis rubicunda - Silvered Leaf Monkey Presbytis cristata 9. Gibbons HYLOBATIDAE - Borneon Gibbon Hylobates muelleri 10. Orangutan PONGIDAE - Borneon Orangutan Pongo pymaeus 11. Bears URSIDAE - Sun Bear Helarctos malayanus 12. Weasels MUSTELIDAE - Yellow-throated Marten Martes fulvigula - Malay Weasel Mustila nudipes - Ferret Badger Melogale orientalis - Malay Badger Mydaus javanensis - Hairy-nosed Otter Lutra sumatrana - Smooth Otter Lutrogale perspicillata - Common Otter Lutra lutra - Oriental Small-clawed Otter Aonycx cinerea 13. Civets VIVERRIDAE - Malay Civet Viverra tangalunga - Banded Linsang Prionodon lisang - Common Palm Civet Paradoxurus hermaphrodilus - Masked Palm Civet Paguma larvata - Binturong Arctitis binturong -

Terrestrial Mammal Community Richness and Temporal Overlap Between Tigers and Other Carnivores in Bukit Barisan Selatan National Park, Sumatra
Animal Biodiversity and Conservation 43.1 (2020) 97 Terrestrial mammal community richness and temporal overlap between tigers and other carnivores in Bukit Barisan Selatan National Park, Sumatra M. L. Allen, M. C. Sibarani, L. Utoyo, M. Krofel Allen, M. L., Sibarani, M. C., Utoyo, L., Krofel, M., 2020. Terrestrial mammal community richness and temporal overlap between tigers and other carnivores in Bukit Barisan Selatan National Park, Sumatra. Animal Biodi- versity and Conservation, 43.1: 97–107, DOI: https://doi.org/10.32800/abc.2020.43.0097 Abstract Terrestrial mammal community richness and temporal overlap between tigers and other carnivores in Bukit Barisan Selatan National Park, Sumatra. Rapid and widespread biodiversity losses around the world make it important to survey and monitor endangered species, especially in biodiversity hotspots. Bukit Barisan Selatan National Park (BBSNP) is one of the largest conserved areas on the island of Sumatra, and is important for the conservation of many threatened species. Sumatran tigers (Panthera tigris sumatrae) are critically endangered and serve as an umbrella species for conservation, but may also affect the activity and distribution of other carnivores. We deployed camera traps for 8 years in an area of Bukit Barisan Selatan National Park (BBSNP) with little human activity to document the local terrestrial mammal community and investigate tiger spatial and temporal overlap with other carnivore species. We detected 39 mammal species including Sumatran tiger and several other threatened mammals. Annual species richness averaged 21.5 (range 19–24) mammals, and re- mained stable over time. The mammal order significantly affected annual detection of species and the number of cameras where a species was detected, while species conservation status did not. -

Movement Patterns of Rats and Treeshrews in Bornean Rainforest Inferred from Mark- Recapture Data
ECOTROPICA 14: 113–120, 2008 © Society for Tropical Ecology MOVEMENT PATTERNS OF RATS AND TREESHREWS IN BORNEAN RAINFOREST INFERRED FROM MARK- RECAPTURE DATA Konstans Wells1*, Maklarin B. Lakim2 & Martin Pfeiffer1 1 Institute of Experimental Ecology, University of Ulm, Albert-Einstein Allee 11, D-89069 Ulm, Germany 2 Sabah Parks, Peti Surat 10626, 88806 Kota Kinabalu, Sabah, Malaysia Abstract. Movement and space-use patterns influence animal population dynamics and interactions with the environment in various ways. In this study, we estimated movement patterns of six rat (Muridae) and four treeshrew (Tupaiidae) species from Borneo based on mark-recapture data in three unlogged and three logged rainforest sites. The treeshrews Tupaia gracilis, T. longipes, and T. tana moved longer straight-line distances than any of the observed rats, with the largest movement distances recorded for T. longipes. The largest mean movement distance for rats was observed for the giant rat Leopoldamys sabanus, while the shortest movements were recorded for the rats Maxomys whiteheadi and Sundamys muelleri. Generally, rats showed a higher frequency of short movements than treeshrews, suggesting that rats are more restricted in their ranging patterns compared with the more exploratory movements of treeshrews. Mixed effects modeling suggests a significant impact of forest type on movement patterns of L. sabanus, and marginal impacts on the movements of N. cremoriventer and T. longipes. Significant sex- and age-specific effects were detected for L. sabanus, N. cremoriventer, M. surifer, M. white- headi, S. muelleri, T. longipes, and T. gracilis. Interacting effects such as forest age and type on movements of L. sabanus, as well as the relatively weak effect of forest type in mixed-effects analysis for T. -
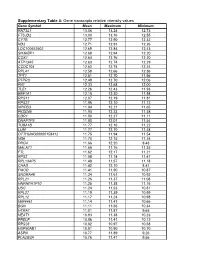
Gene Transcripts Relative Intensity Values
Supplementary Table 4: Gene transcripts relative intensity values Gene Symbol Mean Maximum Minimum RN7SL1 13.06 13.24 12.73 FTSJD2 13.00 13.16 12.55 CYTB 12.77 12.90 12.32 ND2 12.71 12.91 12.26 LOC100652902 12.69 12.84 12.43 SH3KBP1 12.68 12.94 12.20 COX1 12.64 12.76 12.20 ATP13A5 12.63 12.74 12.29 CCDC104 12.62 12.74 12.24 RPL41 12.58 12.66 12.36 TPT1 12.51 12.70 11.96 PTPRO 12.49 12.70 12.06 FN1 12.33 12.68 12.00 TLE1 12.26 12.43 11.93 EEF1A1 12.15 12.30 11.98 RPS11 12.07 12.19 11.81 RPS27 11.96 12.10 11.72 NPIPB3 11.94 12.21 11.65 FKSG49 11.94 12.23 11.58 CDR1 11.90 12.27 11.11 DNAPTP3 11.80 12.07 11.54 TUBA1B 11.77 12.16 11.22 LUM 11.77 12.10 11.48 OTTHUMG00000158412 11.75 11.94 11.54 ND6 11.70 12.15 11.24 PRG4 11.66 12.20 9.43 MALAT1 11.65 11.76 11.35 FTL 11.62 12.17 11.21 RPS2 11.58 11.74 11.47 RPL13AP5 11.48 11.57 11.18 CHAD 11.42 12.10 8.41 FMOD 11.41 11.90 10.87 SNORA48 11.34 11.61 10.92 RPL21 11.26 11.37 11.08 HNRNPA1P10 11.25 11.38 11.16 UBC 11.24 11.53 10.81 RPL27 11.19 11.39 10.99 RPL12 11.17 11.24 10.98 MIR4461 11.14 11.47 10.66 BGN 11.11 11.66 10.34 HTRA1 11.01 11.87 9.65 NEAT1 10.93 11.38 10.23 PRELP 10.86 11.41 10.12 RPS28 10.82 10.97 10.58 HSP90AB1 10.81 10.90 10.70 ASPN 10.77 11.89 9.26 PLA2G2A 10.76 11.47 9.56 H3F3A 10.69 10.86 10.41 EEF1G 10.69 10.94 10.11 C6orf48 10.64 11.02 10.35 UBA52 10.58 10.75 10.08 CTGF 10.57 11.33 9.66 MGP 10.57 11.22 10.09 YBX1 10.56 10.79 10.01 MT1X 10.56 11.89 9.88 NPC2 10.55 10.80 10.12 LAPTM4A 10.51 10.78 10.18 ITM2B 10.51 10.68 10.34 IBSP 10.50 11.50 7.17 NPIPB5 10.50 10.73 10.26 TUBA1A