Sustainable Chemistry, Synthesis and Structure-Activity Relationship Studies of Biologically Important Small Molecules" (2013)
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Aldrich Vapor
Aldrich Vapor Library Listing – 6,611 spectra This library is an ideal tool for investigator using FT-IR to analyze gas phase materials. It contains gas phase spectra collected by Aldrich using a GC-IR interface to ensure chromatographically pure samples. The Aldrich FT-IR Vapor Phase Library contains 6,611 gas phase FT-IR spectra collected by Aldrich Chemical Company using a GC interface. The library includes compound name, molecular formula, CAS (Chemical Abstract Service) registry number, Aldrich catalog number, and page number in the Aldrich Library of FT-IR Spectra, Edition 1, Volume 3, Vapor-Phase. Aldrich Vapor Index Compound Name Index Compound Name 6417 ((1- 3495 (1,2-Dibromoethyl)benzene; Styrene Ethoxycyclopropyl)oxy)trimethylsilane dibromide 2081 (+)-3-(Heptafluorobutyryl)camphor 3494 (1-Bromoethyl)benzene; 1-Phenylethyl 2080 (+)-3-(Trifluoroacetyl)camphor bromide 262 (+)-Camphene; 2,2-Dimethyl-3- 6410 (1-Hydroxyallyl)trimethylsilane methylenebicyclo[2.2.1]heptane 6605 (1-Methyl-2,4-cyclopentadien-1- 2828 (+)-Diisopropyl L-tartrate yl)manganese tricarbonyl 947 (+)-Isomenthol; [1S-(1a,2b,5b)]-2- 6250 (1-Propynyl)benzene; 1-Phenylpropyne Isopropyl-5-methylcyclohexano 2079 (1R)-(+)-3-Bromocamphor, endo- 1230 (+)-Limonene oxide, cis + trans; (+)-1,2- 2077 (1R)-(+)-Camphor; (1R)-(+)-1,7,7- Epoxy-4-isopropenyl-1- Trimethylbicyclo[2.2.1]heptan- 317 (+)-Longifolene; (1S)-8-Methylene- 976 (1R)-(+)-Fenchyl alcohol, endo- 3,3,7-trimethyltricyclo[5.4.0 2074 (1R)-(+)-Nopinone; (1R)-(+)-6,6- 949 (+)-Menthol; [1S-(1a,2b,5a)]-(+)-2- Dimethylbicyclo[3.1.1]heptan-2- -

Title Crystallization of Stereospecific Olefin Copolymers (Special Issue on Physical Chemistry) Author(S) Sakaguchi, Fumio; Kita
Crystallization of Stereospecific Olefin Copolymers (Special Title Issue on Physical Chemistry) Author(s) Sakaguchi, Fumio; Kitamaru, Ryozo; Tsuji, Waichiro Bulletin of the Institute for Chemical Research, Kyoto Citation University (1966), 44(4): 295-315 Issue Date 1966-10-31 URL http://hdl.handle.net/2433/76134 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Crystallization of Stereospecifie Olefin Copolymers Fumio SAKAGUCHI,Ryozo KITAMARU and Waichiro TSUJI* (Tsuji Laboratory) Received August 13, 1966 The stereoregularity of isotactic poly(4-methyl-1-pentene) was characterized and isomorphism phenomena were examined for the copolymeric systems of 4-methyl-1-pentene with several olefins in order to study the crystallization phenomena in these olefin copoly- mers polymerized with stereospecific catalysts. The structural heterogeneity or the fine crystalline structure of poly(4-methyl-1-pentene) could be correlated with its molecular structure by viewing this stereoregular homopolymer as if it were a copolymer. Cocrystallization or isomorphism phenomenon was recognized for the copolymeric systems of 4-methyl-1-pentene with butene-1, pentene-1, decene-1 and 3-methyl-1-butene, while no evidence of the phenomenon was obtained for the copolymeric systems with styrene and propylene. The degree of the isomorphism of those copolymers was discussed with the informations on the crystalline phases obtained from the X-ray study, on the constitution of the copolymeric chains in the amorphous phases obtained from the viscoelastic studies and on the other thermodynamical properties of these systems. INTRODUCTION Many works have been made with regard to the homopolymerization of olefins with stereospecific catalysts, i. e. complex catalysts composed of the combination of organometallic compound and transitional metallic compound. -

Highly Efficient Olefin Isomerization Catalyzed by Metal Hydrides Derives from Dirhodium(Ii) Carboxylates and Catecholborane
HIGHLY EFFICIENT OLEFIN ISOMERIZATION CATALYZED BY METAL HYDRIDES DERIVES FROM DIRHODIUM(II) CARBOXYLATES AND CATECHOLBORANE Gene A. Devora and Michael P. DoyleL' * Department of Chemistry, Trinity University, San Antonio, Texas 78212, USA Abstract. Dirhodium(ll) tetraacetate in combination with catecholborane catalyzes the iso- merization of alkenes and dienes. Effective isomerization occurs at 80-135°C with the use of only 0.1 mol % rhodium acetate. With 2-methyl-1,5-hexadiene the disubstituted double bond is prefer- entially isomerized. In addition, hydrogen transfer hydrogenation occurs with 1,4-cyclohexadienes. The mechanism of these reactions is proposed to involve organoborane addition across a Rh-0 bond which activates the catalyst for isomerization and hydrogenation. INTRODUCTION Catalytic isomerization of alkenes is a characteristic transformation of transition metal hy- drides that often accompanies hydrogenation1 and is one of the most thoroughly studied catalytic reactions.2"4 Compounds of cobalt, nickel, palladium, platinum, rhodium, and ruthenium are effective,2 but other transition metal compounds have also been employed for catalytic isomeriza- tions.2"4 Although the nature of this transformation is dependent on the catalyst, selectivity for alkene isomerization generally favors reactions with monosubstituted ethylenes over di- and tri-sub- stituted ethylenes. In the course of our investigations of the catalytic effectiveness of dirhodium(ll) tetrakis(carboxylates) we have uncovered a useful methodology for the generation of rhodium hydride species that, as we now report, are surprisingly effective for the isomerization of alkenes as well as for hydrogen transfer hydrogenation. MATERIALS AND METHODS Reactions were performed in a round bottom flask equipped with a screw cap that was fitted with a septum for convenient withdrawal of aliquots. -

September 17, 2007
Pre-Feasibility Report M/s. Neogen Chemicals Ltd. 1 1. Introduction M/s. Neogen Chemicals Ltd. is a new unit located at Plot No. Z/96/B SEZ Dahej, District: Bharuch, Gujarat. Now, the unit proposes to manufacture different type of synthetic organic dyes and pesticide products at above sited address. 2. Cost of Project Cost of existing project is 55 crore &, out of which 5 crore will be used for Environment Management System. 3. Production Capacity Production capacity is prescribe below: List of Products Sr. Name of Products Quantity No. (MT/Year) (MT/month) 1 Bromination and Chlorination of Alcohols 1.1. Ethyl Bromide 3500 291.67 1.2. n-Propyl Bromide 1.3. Iso Propyl Bromide 1.4. n-Butyl Bromide 1.5. Iso Butyl Bromide 1.6. Sec-Butyl Bromide 1.7. n-Hexyl Bromide 1.8. n-Heptyl Bromide 1.9. n-Octyl Bromide 1.10. n-Decyl Bromide 1.11. Lauryl Bromide 1.12. Cetyl Bromide 1.13. Myristyl Bromide 1.14. Stearyl Bromide 1.15. 1,2 Dibromo Ethane 1.16. 1,3 Dibromo Propane 1.17. 1,4 Dibromo Butane 1.18. 1,5 Dibromo pentane M/s. Neogen Chemicals Ltd. 2 Sr. Name of Products Quantity No. (MT/Year) (MT/month) 1.19. 1,6 Dibromo Hexane 1.20. 1 Chloro 2 Ethyl Hexane 1.21. 6 Chloro 1 Hexanol 1.22. 3 Chloro Propanol 1.23. 1,6 Dichloro Hexane 1.24. Cyclo Propyl Methyl Bromide 1.25. Cyclo Pentyl Bromide 1.26. Cyclo Pentyl Chloride 2. Bromination of Organic Acids and Esterification thereof 2.1. -

' United "States Patent Office
Patented on. 20, 1.942 I . 2,299,411 ' UNITED "STATES PATENT OFFICE CATALYZED HYDROBROMINATION OF UN SATURATED ORGANIC COMPOUNDS Fredericlr Rust and William E. Vaughan, ' Berkeley, Calif., assignors. to Shell Develop ment Company, San Francisco, Calif., a corpo ration of Delaware No Drawing. 4 Application August 25, 1941, Serial No. 408,212 - 9'Claims. (01. 260-663) ' This invention relates to an improved process for the hydrobromination of unsaturated organic hydes and metal alkyls which tend to initiate I compounds, and more particularly to improve the reaction chains. v ments in the method of controlling the addition It is known that the presence of peroxide: orv of hydrogen bromide to unsymmetrical organic of peroxide-forming compounds in unsaturated compounds containing at least one ole?nic or organic compounds, e. g. unsaturated hydrocar acetylenic linkage to produce addition products bons, is undesirable. For example, organic per of a predetermined character. oxides, when present even in relatively small con It is known that hydrogen halides may be centrations, tend to catalyze the polymerization of a large number of unsaturated hydrocarbons, . added to unsaturated hydrocarbons and to var 10 ious unsaturated derivatives thereof. In fact, and particularly diole?ns. As to the “abnormal” in 1870 Markowniko? stated that “if an addition ofhydrogen bromide to unsaturates by metrical hydrocarbon combines with a halogen effecting the reaction under the deliberate in acid, the halogen adds to the carbon atom with ?uence, of light, and particularly of ultra-violet fewer hydrogen atoms, i. e. to the carbon atom radiations having wave-lengths of below about which is more'under the in?uence of other car' 2900 to 3000 Angstrom units, such processes ne bon atoms.” The same investigator further de cessitate the use of special equipment, such as termined that when a hydrogen halide is added reaction vessels provided with or containing to a halogenated unsaturated compound such as lamps made of quartz Or other suitable mate rials, e. -

Chemical Compatibility Chart X
Chemical Compatibility Chart Below is a chart adapted from the CRC Laboratory Handbook, which groups various chemicals in to 23 groups with examples and incompatible chemical groups. This chart is by no means complete but it will aid in making decisions about storage. For more complete information please refer to the MSDS for the specific chemical. Examples of each group can be found on the next pages. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Monomers Polymerizable Esters Alcohols, Glycols, Glycol Ether Amines and Alkanolamines Halogenated Compounds Aldehydes Acetaldehyde Saturated Hydrocar Aromatic Hydrocarbons Acid Anhydrides Alkylene Oxides Inorganic Acids Petrolium Oils Organic Acids Cyanohydrins Phosphorus Ammonia Group Halogens Ketones Caustics Phenols Nitriles Olefins Ethers Number/Chemical Esters Type bons Inorganic 1 x x x x x x x x x x x x x x x x x Acids 2 Organic Acids x x x x x x x x x x 3 Caustics x x x x x x x x x x x x x Amines and 4 x x x x x x x x x x x x Alkanolamines Halogenated 5 x x x x x x Compounds Alcohols, 6 Glycols, Glycol x x x x x x Ether Aldehydes 7 x x x x x x x x x x x x Acetaldehyde 8 Ketones x x x x x x Saturated 9 x Hydrocarbons Aromatic 10 x x Hydrocarbons 11 Olefins x x x 12 Petrolium Oils x 13 Esters x x x x x Monomers 14 Polymerizable x x x x x x x x x x x x Esters 15 Phenols x x x x x x x Alkylene 16 x x x x x x x x x x x x Oxides 17 Cyanohydrins x x x x x x x x x 18 Nitriles x x x x x x 19 Ammonia x x x x x x x x x x x 20 Halogens x x x x x x x x x x x x x x 21 Ethers x x x 22 Phosphorus x x x x Acid 23 x x x x x x x x x x Anhydrides X - Indicates chemicals that are incompatible and should not be stored together. -
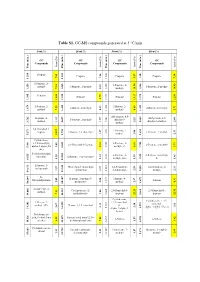
Table S1. GC-MS Compounds Generated at 5 °C/Min
Table S1. GC-MS compounds generated at 5 °C/min. 0 wt.% 10 wt.% 30 wt.% 50 wt.% GC GC GC GC Compounds Compounds Compounds Compounds Area (%) Area (%) Area (%) Area (%) Time (min.) (min.) Time Time (min.) (min.) Time (min.) Time (min.) Time Propene Propene Propene Propene 7.42 7.42 1.26 5.47 1.20 7.38 1.200 1.200 1.200 1.214 1.214 1-Propene, 2- 1-Propene, 2- methyl- 1-Propene, 2-methyl- 1-Propene, 2-methyl- 7.34 7.34 2.41 methyl- 1.245 1.245 1.245 1.259 1.259 10.54 1.246 14.90 Pentane Pentane Pentane Pentane 3.79 3.79 1.375 1.375 1.395 1.395 11.81 1.381 13.17 1.375 12.83 1-Pentene, 2- 2-Butene, 2- 2-Butene, 2-methyl- 2-Butene, 2-methyl- methyl- 7.25 0.93 methyl- 4.65 5.71 1.426 1.426 1.420 1.420 1.679 1.679 1H-Pyrazole, 4,5- Heptane, 4- 1H-Pyrazole, 4,5- 1-Pentene, 2-methyl- dihydro-5- methyl- 1.92 1.57 2.25 dihydro-5-methyl- 2.44 1.659 1.659 1.562 1.569 3.762 3.762 methyl- 2,4-Dimethyl-1- 1-Pentene, 2- heptene 2-Butene, 2,3-dimethyl- 1-Pentene, 2-methyl- 1.27 1.27 methyl- 3.08 2.33 1.653 1.653 1.653 5.671 5.671 34.57 1.750 Cyclohexane, 1,3,5-trimethyl-, 2-Pentene, 3- 2,4-Dimethyl-1-heptene 2-Pentene, 2-methyl- alpha.,3.alpha.,5.b 2.03 9.11 methyl-, €- 6.11 5.73 5.605 5.605 1.743 1.744 5.981 5.981 eta.)- 5-Aminoisoxazole 2-Pentene, 3- 2-Pentene, 3-methyl-, Spermine Ethanone, 1-cyclopentyl- 1.55 1.55 1.32 methyl-, (Z)- 1.59 €- 1.46 1.828 1.828 1.828 6.925 6.925 10.560 Ethanone, 1- 1R,2c,3t,4t-Tetramethyl- 1,3-Pentadiene, 1,4-Hexadiene, 5- cyclopentyl- 3.57 3.57 cyclohexan 0.97 2,3-dimethyl- 2.28 methyl- 2.31 2.584 2.584 2.585 10.613 10.613 10.638 N- Heptane, 2-methyl-3- Heptane, 4- Methylallylamine Toluene 2.49 2.49 methylene- 2.50 methyl- 2.97 4.31 3.697 3.697 3.697 14.047 14.047 10.684 10.684 2-Undecene, 4- Cyclopentane, (2- 2,4-Dimethyl-1- 2,4-Dimethyl-1- methyl- 7.81 7.81 methylbutyl)- 1.00 heptene heptene 5.605 5.605 22.29 22.29 5.605 12.95 14.086 14.086 14.163 Cyclohexane, Cyclohexane, 1,3,5- 2-Decene, 7- 1,3,5-trimethyl-, trimethyl- methyl-, (Z)- Hexane, 2,3,4-trimethyl- (1. -

Linear Alpha-Olefins (681.5030)
IHS Chemical Chemical Economics Handbook Linear alpha-Olefins (681.5030) by Elvira O. Camara Greiner with Yoshio Inoguchi Sample Report from 2010 November 2010 ihs.com/chemical November 2010 LINEAR ALPHA-OLEFINS Olefins 681.5030 B Page 2 The information provided in this publication has been obtained from a variety of sources which SRI Consulting believes to be reliable. SRI Consulting makes no warranties as to the accuracy completeness or correctness of the information in this publication. Consequently SRI Consulting will not be liable for any technical inaccuracies typographical errors or omissions contained in this publication. This publication is provided without warranties of any kind either express or implied including but not limited to implied warranties of merchantability fitness for a particular purpose or non-infringement. IN NO EVENT WILL SRI CONSULTING BE LIABLE FOR ANY INCIDENTAL CONSEQUENTIAL OR INDIRECT DAMAGES (INCLUDING BUT NOT LIMITED TO DAMAGES FOR LOSS OF PROFITS BUSINESS INTERRUPTION OR THE LIKE) ARISING OUT OF THE USE OF THIS PUBLICATION EVEN IF IT WAS NOTIFIED ABOUT THE POSSIBILITY OF SUCH DAMAGES. BECAUSE SOME STATES DO NOT ALLOW THE EXCLUSION OR LIMITATION OF LIABILITY FOR CONSEQUENTIAL OR INCIDENTAL DAMAGES THE ABOVE LIMITATION MAY NOT APPLY TO YOU. IN SUCH STATES SRI CONSULTING’S LIABILITY IS LIMITED TO THE MAXIMUM EXTENT PERMITTED BY SUCH LAW. Certain statements in this publication are projections or other forward-looking statements. Any such statements contained herein are based upon SRI Consulting’s current knowledge and assumptions about future events including without limitation anticipated levels of global demand and supply expected costs trade patterns and general economic political and marketing conditions. -

Ionic Liquid*
Pure Appl. Chem., Vol. 83, No. 7, pp. 1391–1406, 2011. doi:10.1351/PAC-CON-10-12-03 © 2011 IUPAC, Publication date (Web): 10 June 2011 Oligomerisation of linear 1-olefins using a chlorogallate(III) ionic liquid* Martin P. Atkins1, Kenneth R. Seddon2,‡, and Małgorzata Swadźba-Kwaśny2,‡ 1PETRONAS Research Sdn Bhd, Lot 3288&3289, Off Jalan Ayer Itam, Kawasan Institusi Bangi, 43000, Kajang, Malaysia; 2QUILL, The Queen’s University of Belfast, Belfast, BT9 5AG, UK Abstract: A Lewis acidic chlorogallate(III) ionic liquid, 1-ethyl-3-methylimidazolium hepta - chlorodigallate(III), [C2mim][Ga2Cl7], was successfully used to oligomerise 1-pentene. The influence of temperature, time, catalyst concentration, and stirring rate on conversion and product distribution was modelled using a design of experiment (DoE) approach (chemo - metrics). The process was optimised for lubricant base oils production; the C20–C50 fraction (where Cn indicates the number of carbons in the oligomer) was maximised, while the heav- ier oligomer fraction (>C50) was minimised. Keywords: 1-alkenes; chemometrics; ionic liquids; linear terminal olefins; 1-olefins; oligomerisation. INTRODUCTION Oligomerisation and ionic liquids Polyalphaolefins (PAOs), used as a base for synthetic automotive lubricants (Group 4 base stock by API classification) [1], are traditionally made by oligomerisation of 1-decene to C20–C50 blend (where Cn indicates the number of carbons in the oligomer) [2] using Ziegler–Natta or Friedel–Crafts catalysts [3]. The carbon number is important, as the pour point of higher polymers (>C50) is too high, whilst the lower cut (<C20) is too volatile. 1-Decene is manufactured in a very selective manner from ethene [2]. -

Synthetic Applications of Organoboranes
ú 7lt) f SYNTHETIC APPLICATIONS OF ORGANOBORANES A Thesis Presented for the Degree of Doctor of Philosophy in The Uníversity of Adelaide by Roger Murphy, B.Sc. (Hons.) Department of Organíc Chemistry L976. CONTENTS Page SUMMARY (f) STATEMENT (fií) PUBL ICATIONS (iv) ACKNOI^JLEDGEMENTS (v) CHAPTER 1. Synthesis of Products Isolated from the Hydroboration-Cyanidation of Unsaturated Terpenes. / 1.1 Introduction. 1 L.2 Synthesis of Products Derived from L2 Geraniol. 1.3 Synthesis of Products Derived from L7 LÍnalyl AceËate. L.4 SynthesÍs of Products Derived from 22 Myrcene. CHAPTER 2. Cyanidation of Dialkyìboranes and Borinic Acids and Esters. 2.L Introduction. 43 2.2 Cyanidation of Dialkylboranes. 50 2.3 Cyanidation of BorÍnic Acíds and EsËers. 65 CHAPTER 3. Silver(I) 0xidation of Organoboranes. 3.1 Introduction. 70 3.2 Cycl-izatlon of Díenes vía Intramolecular 76 Alkyl Coupling. 3.3 AÈtenpted Reduction of Intemedl-ates 84 Obtaíned by Reaction of Organoboranes with Alkalíne SÍ1ver nltrate. CHAPTER 4. Asymmetric Induction by Hydroboration with Optical ly Active Dial llyl boranes. 4.L Introductlon. 88 4.2 AËterpted Resolution of (t)-414,6- 96 Trfnethylcaprolactam and (t)-4 16 16- Tr lmethylcapro lactam. 4.3 Attempted Resolutíon of (1)-31515- 100 Trinethylcyclohexanone . 4.4 Asynrmetric Hydroboratfon of 6r7-Dihydro- 110 416 r6-trTmethyl-5H-azepínone and 3r5,5- TrirneËhylcyclohex-2-enone (Isophorone) . CHAPTER 5. Experimental. 5.1 General. 118 5.2 I^Iork described in chapter 1. L2T 5.3 Ilork described in chapter 2. 163 5.4 Work descríbed ín chapËer 3. 178 5.5 lJork described Ín chapter 4. -

Light Linear Alpha Olefin Market Study
IHS CHEMICAL Light Linear Alpha Olefin Market Study Special Report Prospectus IHS CHEMICAL Contents Contents ..................................................................................................................................... 2 Introduction ................................................................................................................................ 3 Study Scope ............................................................................................................................... 5 Key Questions Addressed in the Study ...................................................................................... 7 Deliverables ............................................................................................................................... 8 Proposed Table of Contents ...................................................................................................... 9 Methodology ............................................................................................................................ 11 Study Team .............................................................................................................................. 15 Qualifications............................................................................................................................ 18 About IHS Chemical ................................................................................................................. 21 About IHS ................................................................................................................................