Asgard Archaea Capable of Anaerobic Hydrocarbon Cycling
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Syntrophy Hypothesis for the Origin of Eukaryotes Revisited Purificación López-García, David Moreira
The Syntrophy hypothesis for the origin of eukaryotes revisited Purificación López-García, David Moreira To cite this version: Purificación López-García, David Moreira. The Syntrophy hypothesis for the origin of eukaryotes revisited. Nature Microbiology, Nature Publishing Group, 2020, 5 (5), pp.655-667. 10.1038/s41564- 020-0710-4. hal-02988531 HAL Id: hal-02988531 https://hal.archives-ouvertes.fr/hal-02988531 Submitted on 3 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 2 Perspectives 3 4 5 6 The Syntrophy hypothesis for the origin of eukaryotes revisited 7 8 Purificación López-García1 and David Moreira1 9 10 1 Ecologie Systématique Evolution, CNRS, Université Paris-Saclay, AgroParisTech, Orsay, France 11 12 13 14 *Correspondence to: [email protected] 15 16 17 18 19 1 20 The discovery of Asgard archaea, phylogenetically closer to eukaryotes than other archaea, together with 21 improved knowledge of microbial ecology impose new constraints on emerging models for the origin of the 22 eukaryotic cell (eukaryogenesis). Long-held views are metamorphosing in favor of symbiogenetic models 23 based on metabolic interactions between archaea and bacteria. These include the classical Searcy’s and 24 hydrogen hypothesis, and the more recent Reverse Flow and Entangle-Engulf-Enslave (E3) models. -

Marsarchaeota Are an Aerobic Archaeal Lineage Abundant in Geothermal Iron Oxide Microbial Mats
Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats Authors: Zackary J. Jay, Jacob P. Beam, Mansur Dlakic, Douglas B. Rusch, Mark A. Kozubal, and William P. Inskeep This is a postprint of an article that originally appeared in Nature Microbiology on May 14, 2018. The final version can be found at https://dx.doi.org/10.1038/s41564-018-0163-1. Jay, Zackary J. , Jacob P. Beam, Mensur Dlakic, Douglas B. Rusch, Mark A. Kozubal, and William P. Inskeep. "Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats." Nature Microbiology 3, no. 6 (May 2018): 732-740. DOI: 10.1038/ s41564-018-0163-1. Made available through Montana State University’s ScholarWorks scholarworks.montana.edu Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats Zackary J. Jay1,4,7, Jacob P. Beam1,5,7, Mensur Dlakić2, Douglas B. Rusch3, Mark A. Kozubal1,6 and William P. Inskeep 1* The discovery of archaeal lineages is critical to our understanding of the universal tree of life and evolutionary history of the Earth. Geochemically diverse thermal environments in Yellowstone National Park provide unprecedented opportunities for studying archaea in habitats that may represent analogues of early Earth. Here, we report the discovery and character- ization of a phylum-level archaeal lineage proposed and herein referred to as the ‘Marsarchaeota’, after the red planet. The Marsarchaeota contains at least two major subgroups prevalent in acidic, microaerobic geothermal Fe(III) oxide microbial mats across a temperature range from ~50–80 °C. Metagenomics, single-cell sequencing, enrichment culturing and in situ transcrip- tional analyses reveal their biogeochemical role as facultative aerobic chemoorganotrophs that may also mediate the reduction of Fe(III). -

Phylogenomics Provides Robust Support for a Two-Domains Tree of Life
ARTICLES https://doi.org/10.1038/s41559-019-1040-x Phylogenomics provides robust support for a two-domains tree of life Tom A. Williams! !1*, Cymon J. Cox! !2, Peter G. Foster3, Gergely J. Szöllősi4,5,6 and T. Martin Embley7* Hypotheses about the origin of eukaryotic cells are classically framed within the context of a universal ‘tree of life’ based on conserved core genes. Vigorous ongoing debate about eukaryote origins is based on assertions that the topology of the tree of life depends on the taxa included and the choice and quality of genomic data analysed. Here we have reanalysed the evidence underpinning those claims and apply more data to the question by using supertree and coalescent methods to interrogate >3,000 gene families in archaea and eukaryotes. We find that eukaryotes consistently originate from within the archaea in a two-domains tree when due consideration is given to the fit between model and data. Our analyses support a close relation- ship between eukaryotes and Asgard archaea and identify the Heimdallarchaeota as the current best candidate for the closest archaeal relatives of the eukaryotic nuclear lineage. urrent hypotheses about eukaryotic origins generally pro- Indeed, it has previously been suggested that it is the 3D tree, rather pose at least two partners in that process: a bacterial endo- than the 2D tree, that is an artefact of long-branch attraction5,9–11, symbiont that became the mitochondrion and a host cell for both because analyses under better-fitting models have recovered C 1–4 that endosymbiosis . The identity of the host has been informed a 2D tree but also because the 3D topology is one in which the two by analyses of conserved genes for the transcription and transla- longest branches in the tree of life—the stems leading to bacteria and tion machinery that are considered essential for cellular life5. -

ARCHAEAL EVOLUTION Evolutionary Insights from the Vikings
RESEARCH HIGHLIGHTS Nature Reviews Microbiology | Published online 16 Jan 2017; doi:10.1038/nrmicro.2016.198 ARCHAEAL EVOLUTION Evolutionary insights from the Vikings The emergence of the eukaryotic cell ASGARD — after the invisible the closest known homologue of during evolution gave rise to all com- ‘Gods of Asgard’ in Norse mythology. eukaryotic epsilon DNA polymerases primordial plex life forms on Earth, including The superphylum consists of the identified thus far. eukaryotic multicellular organisms such as ani- previously identified Lokiarchaeota Members of the ASGARD super- mals, plants and fungi. However, the and Thorarchaeota phyla, and the phylum were particularly enriched vesicular and origin of eukaryotes and their char- newly identified Odinarchaeota and for eukaryotic signature proteins trafficking acteristic structural complexity has Heimdallarchaeota phyla. Using that are involved in intracellular components remained a mystery. The most recent phylogenomics, they discovered trafficking and secretion. Several are derived insights into eukaryogenesis support a strong phylogenetic association proteins contained domain signatures the endosymbiotic theory, which between ASGARD lineages and of eukaryotic transport protein from our proposes that the first eukaryotic eukaryotes that placed the eukaryote particle (TRAPP) complexes, which archaeal cell arose from archaea through the lineage in close proximity to the are involved in transport from the ancestor acquisition of an alphaproteobacterial ASGARD superphylum. endoplasmic -

Two Domains of Life, Not Three?
Asgard Archaea Two domains of life, not three? A picture of visual interpretation of the Wagner’s opera Das Rheingold, a part of Richard Wagner’s Der Ring des Nibelungen Loki's Castle is a field of five active hydrothermal vents in the mid-Atlantic Ocean, located at 73 degrees north on the Mid-Atlantic Ridge between Greenland and Norway at a depth of 2,352 metres The vents were discovered in 2008 by a multinational scientific expedition of the university of Bergen, and are the most northerly black smokers to date. The five active chimneys of Loki's Castle are venting water as hot as 300 °C and sit on a vast mound of sulfide minerals. The vent field was given the name Loki's Castle as its shape reminded its discoverers of a fantasy castle. The reference is to the ancient Norse god of trickery, Loki. The top three feet (1 m) of a vent chimney almost 40 feet (12 m) tall at Loki's Castle in mid-July 2008. Visible at left is the arm of a remotely operated vehicle, reaching in to take fluid samples. Loki’s Castle Wents Loki's Castle - a field of five active hydrothermal vents in the mid-Atlantic Ocean - at 73 degrees north on the Mid-Atlantic Ridge - at a depth of 2,352 meters In Norse mythology, Loki is a cunning trickster who has the ability to change his shape and sex. Loki is represented as the companion of the great gods Odin and Thor. Preliminary observations have shown the warm area around the Loki's Castle vents to be alive with diverse and apparently unique microorganisms, unlike vent communities observed elsewhere. -
Lokiarchaeota: Biologists Discover 'Missing Link' Microorganism
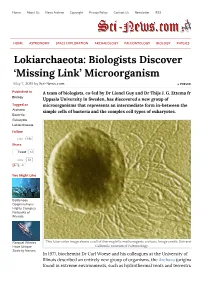
Home About Us News Archive Copyright Privacy Policy Contact Us Newsletter RSS HOME ASTRONOMY SPACE EXPLORATION ARCHAEOLOGY PALEONTOLOGY BIOLOGY PHYSICS Lokiarchaeota: Biologists Discover ‘Missing Link’ Microorganism May 7, 2015 by Sci-News.com « PREVIOUS Published in A team of biologists, co-led by Dr Lionel Guy and Dr Thijs J. G. Ettema from Biology Uppsala University in Sweden, has discovered a new group of Tagged as microorganisms that represents an intermediate form in-between the Archaea simple cells of bacteria and the complex cell types of eukaryotes. Bacteria Eukaryote Lokiarchaeota Follow Like 16k Share Tweet 12 Like 58 41 You Might Like Bottlenose Dolphins Form Highly Complex Networks of Friends Rorqual Whales This false-color image shows a cell of thermophilic methanogenic archaea. Image credit: University of Have Unique California Museum of Paleontology. Stretchy Nerves In 1977, biochemist Dr Carl Woese and his colleagues at the University of Illinois described an entirely new group of organisms, the Archaea (originally found in extreme environments, such as hydrothermal vents and terrestrial hot springs). The scientists were studying relationships among the prokaryotes using DNA Extinction of sequences, and found that Archaea have distinct molecular characteristics World’s Largest separating them from bacteria as well as from eukaryotes. They proposed that Herbivores May Lead to Empty life can be divided into three domains: Eukaryota, Eubacteria, and Landscapes, Say Archaebacteria. Researchers Despite that archaeal cells were simple and small like bacteria, scientists found that Archaea were more closely related to organisms with complex cell types, a group collectively known as ‘eukaryotes.’ This observation has puzzled Sichuan Bush biologists for years. -

Helarchaeota and Co-Occurring Sulfate-Reducing Bacteria in Subseafloor
www.nature.com/ismecomms ARTICLE OPEN Helarchaeota and co-occurring sulfate-reducing bacteria in subseafloor sediments from the Costa Rica Margin ✉ Rui Zhao1 and Jennifer F. Biddle 1 © The Author(s) 2021 Deep sediments host many archaeal lineages, including the Asgard superphylum which contains lineages predicted to require syntrophic partnerships. Our knowledge about sedimentary archaeal diversity and their metabolic pathways and syntrophic partners is still very limited. We present here new genomes of Helarchaeota and the co-occurring sulfate-reducing bacteria (SRB) recovered from organic-rich sediments off Costa Rica Margin. Phylogenetic analyses revealed three new metagenome-assembled genomes (MAGs) affiliating with Helarchaeota, each of which has three variants of the methyl-CoM reductase-like (MCR-like) complex that may enable them to oxidize short-chain alkanes anaerobically. These Helarchaeota have no multi-heme cytochromes but have Group 3b and Group 3c [NiFe] hydrogenases, and formate dehydrogenase, and therefore have the capacity to transfer the reducing equivalents (in the forms of hydrogen and formate) generated from alkane oxidation to external partners. We also recovered five MAGs of SRB affiliated with the class of Desulfobacteria, two of which showed relative abundances (represented by genome coverages) positively correlated with those of the three Helarchaeota. Genome analysis suggested that these SRB bacteria have the capacity of H2 and formate utilization and could facilitate electron transfers from other organisms by means of these reduced substances. Their co-occurrence and metabolic features suggest that Helarchaeota may metabolize synergistically with some SRB, and together exert an important influence on the carbon cycle by mitigating the hydrocarbon emission from sediments to the overlying ocean. -

Differential Depth Distribution of Microbial Function and Putative Symbionts Through Sediment-Hosted Aquifers in the Deep Terrestrial Subsurface
ARTICLES https://doi.org/10.1038/s41564-017-0098-y Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface Alexander J. Probst1,5,7, Bethany Ladd2,7, Jessica K. Jarett3, David E. Geller-McGrath1, Christian M. K. Sieber1,3, Joanne B. Emerson1,6, Karthik Anantharaman1, Brian C. Thomas1, Rex R. Malmstrom3, Michaela Stieglmeier4, Andreas Klingl4, Tanja Woyke 3, M. Cathryn Ryan 2* and Jillian F. Banfield 1* An enormous diversity of previously unknown bacteria and archaea has been discovered recently, yet their functional capacities and distributions in the terrestrial subsurface remain uncertain. Here, we continually sampled a CO2-driven geyser (Colorado Plateau, Utah, USA) over its 5-day eruption cycle to test the hypothesis that stratified, sandstone-hosted aquifers sampled over three phases of the eruption cycle have microbial communities that differ both in membership and function. Genome- resolved metagenomics, single-cell genomics and geochemical analyses confirmed this hypothesis and linked microorganisms to groundwater compositions from different depths. Autotrophic Candidatus “Altiarchaeum sp.” and phylogenetically deep- branching nanoarchaea dominate the deepest groundwater. A nanoarchaeon with limited metabolic capacity is inferred to be a potential symbiont of the Ca. “Altiarchaeum”. Candidate Phyla Radiation bacteria are also present in the deepest groundwater and they are relatively abundant in water from intermediate depths. During the recovery phase of the geyser, microaerophilic Fe- and S-oxidizers have high in situ genome replication rates. Autotrophic Sulfurimonas sustained by aerobic sulfide oxidation and with the capacity for N2 fixation dominate the shallow aquifer. Overall, 104 different phylum-level lineages are present in water from these subsurface environments, with uncultivated archaea and bacteria partitioned to the deeper subsurface. -

Origin and Evolution of Eukaryotic Compartmentalization
TESI DOCTORAL UPF 2016 Origin and evolution of eukaryotic compartmentalization Alexandros Pittis Director Dr. Toni Gabaldon´ Comparative Genomics Group Bioinformatics and Genomics Department Centre for Genomic Regulation (CRG) To my father Stavros for the PhD he never did Acknowledgments Looking back at the years of my PhD in the Comparative Genomics group at CRG, I feel it was equally a process of scientific, as well as personal development. All this time I was very privileged to be surrounded by people that offered me their generous support in both fronts. And they were available in the very moments that I was fighting more myself than the unsolvable (anyway) comparative genomics puzzles. I hope that in the future I will have many times the chance to express them my appreciation, way beyond these few words. First, to Toni, my supervisor, for all his support, and patience, and confidence, and respect, especially at the moments that things did not seem that promising. He offered me freedom, to try, to think, to fail, to learn, to achieve that few enjoy during their PhD, and I am very thankful to him. Then, to my good friends and colleagues, those that I found in the group already, and others that joined after me. A very special thanks to Marinita and Jaime, for all their valuable time, and guidance and paradigm, which nevertheless I never managed to follow. They have both marked my phylogenomics path so far and I cannot escape. To Les and Salvi, my fellow students and pals at the time for all that we shared; to Gab and Fran and Dam, for -

Spatial Separation of Ribosomes and DNA in Asgard Archaeal Cells ✉ ✉ Burak Avcı 1,2 , Jakob Brandt3, Dikla Nachmias4, Natalie Elia4, Mads Albertsen 3, Thijs J
www.nature.com/ismej BRIEF COMMUNICATION OPEN Spatial separation of ribosomes and DNA in Asgard archaeal cells ✉ ✉ Burak Avcı 1,2 , Jakob Brandt3, Dikla Nachmias4, Natalie Elia4, Mads Albertsen 3, Thijs J. G. Ettema 2, Andreas Schramm1,5 and Kasper Urup Kjeldsen1 © The Author(s) 2021 The origin of the eukaryotic cell is a major open question in biology. Asgard archaea are the closest known prokaryotic relatives of eukaryotes, and their genomes encode various eukaryotic signature proteins, indicating some elements of cellular complexity prior to the emergence of the first eukaryotic cell. Yet, microscopic evidence to demonstrate the cellular structure of uncultivated Asgard archaea in the environment is thus far lacking. We used primer-free sequencing to retrieve 715 almost full-length Loki- and Heimdallarchaeota 16S rRNA sequences and designed novel oligonucleotide probes to visualize their cells in marine sediments (Aarhus Bay, Denmark) using catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH). Super-resolution microscopy revealed 1–2 µm large, coccoid cells, sometimes occurring as aggregates. Remarkably, the DNA staining was spatially separated from ribosome-originated FISH signals by 50–280 nm. This suggests that the genomic material is condensed and spatially distinct in a particular location and could indicate compartmentalization or membrane invagination in Asgard archaeal cells. The ISME Journal; https://doi.org/10.1038/s41396-021-01098-3 INTRODUCTION cells in marine sediments [8] yet methodical limitations did not The origin of the eukaryotic cell is a major unresolved puzzle in allow to discern single cells and to draw any in-depth conclusion the history of life. -

Archaea and the Origin of Eukaryotes
REVIEWS Archaea and the origin of eukaryotes Laura Eme, Anja Spang, Jonathan Lombard, Courtney W. Stairs and Thijs J. G. Ettema Abstract | Woese and Fox’s 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor. Sister groups Two descendants that split The pioneering work by Carl Woese and colleagues In this Review, we discuss how culture- independent from the same node; the revealed that all cellular life could be divided into three genomics has transformed our understanding of descendants are each other’s major evolutionary lines (also called domains): the archaeal diversity and how this has influenced our closest relative. -

UNIVERSITY of EMBU EDWARD NDERITU KARANJA Phd 2020
UNIVERSITY OF EMBU EDWARD NDERITU KARANJA PhD 2020 MICROBIAL COMMUNITY DIVERSITY AND STRUCTURE WITHIN ORGANIC AND CONVENTIONAL FARMING SYSTEMS IN CENTRAL HIGHLANDS OF KENYA EDWARD NDERITU KARANJA (MSc) A THESIS SUBMITTED IN PARTIAL FULFILLMENT FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN APPLIED MICROBIOLOGY IN THE UNIVERSITY OF EMBU NOVEMBER, 2020 DECLARATION This thesis is my original work and has not been presented for a degree in any other University Signature……………………………. Date………….……….. Edward Nderitu Karanja Department of Biological Science B801/147/2015 This thesis has been submitted for examination with our approval as the University Supervisors Signature……………………………. Date………….………. Prof. Romano Mwirichia Department of Biological science University of Embu (UoEm), Kenya Signature………. …………………………. Date………….…………. Dr. Andreas Fliessbach Department of Soil Science Research Institute of Organic Agriculture - FIBL, Switzerland i DEDICATION I dedicated to my family; my wife Anne Kelly Kambura, my children; Shawn Karanja, Melissa Wangithi, Joseph Munyuithia, Shayne Koome and Ann Wanjiku, my parents; Mr. Samuel Karanja and Mrs. Agnes Wangithi, my siblings, Ruth Wairimu, Juliet Muthoni, Alex Ngochi, James Karuma and Nelly Njoki. I appreciate the support you have accorded me during my studies. Your inspiration and backing in this journey made it easier to manage all challenges encountered. ii ACKNOWLEDGEMENT I express gratitude toward Almighty God for his mercies from the beginning of this long and thought-provoking journey. This was conducted in the framework of long-term systems comparison program, with financial support from Biovision Foundation, Coop Sustainability Fund, Liechtenstein Development Service (LED) and the Swiss Agency for Development and Cooperation (SDC). I acknowledge icipe core funding for the kind contribution provided by UK-Aid from UK Government, Swedish International Development Cooperation Agency, Swiss Agency for Development and Cooperation, Federal Democratic Republic of Ethiopia and the Kenyan Government.