Origin and Evolution of Eukaryotic Compartmentalization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pinpointing the Origin of Mitochondria Zhang Wang Hanchuan, Hubei
Pinpointing the origin of mitochondria Zhang Wang Hanchuan, Hubei, China B.S., Wuhan University, 2009 A Dissertation presented to the Graduate Faculty of the University of Virginia in Candidacy for the Degree of Doctor of Philosophy Department of Biology University of Virginia August, 2014 ii Abstract The explosive growth of genomic data presents both opportunities and challenges for the study of evolutionary biology, ecology and diversity. Genome-scale phylogenetic analysis (known as phylogenomics) has demonstrated its power in resolving the evolutionary tree of life and deciphering various fascinating questions regarding the origin and evolution of earth’s contemporary organisms. One of the most fundamental events in the earth’s history of life regards the origin of mitochondria. Overwhelming evidence supports the endosymbiotic theory that mitochondria originated once from a free-living α-proteobacterium that was engulfed by its host probably 2 billion years ago. However, its exact position in the tree of life remains highly debated. In particular, systematic errors including sparse taxonomic sampling, high evolutionary rate and sequence composition bias have long plagued the mitochondrial phylogenetics. This dissertation employs an integrated phylogenomic approach toward pinpointing the origin of mitochondria. By strategically sequencing 18 phylogenetically novel α-proteobacterial genomes, using a set of “well-behaved” phylogenetic markers with lower evolutionary rates and less composition bias, and applying more realistic phylogenetic models that better account for the systematic errors, the presented phylogenomic study for the first time placed the mitochondria unequivocally within the Rickettsiales order of α- proteobacteria, as a sister clade to the Rickettsiaceae and Anaplasmataceae families, all subtended by the Holosporaceae family. -

Two Domains of Life, Not Three?
Asgard Archaea Two domains of life, not three? A picture of visual interpretation of the Wagner’s opera Das Rheingold, a part of Richard Wagner’s Der Ring des Nibelungen Loki's Castle is a field of five active hydrothermal vents in the mid-Atlantic Ocean, located at 73 degrees north on the Mid-Atlantic Ridge between Greenland and Norway at a depth of 2,352 metres The vents were discovered in 2008 by a multinational scientific expedition of the university of Bergen, and are the most northerly black smokers to date. The five active chimneys of Loki's Castle are venting water as hot as 300 °C and sit on a vast mound of sulfide minerals. The vent field was given the name Loki's Castle as its shape reminded its discoverers of a fantasy castle. The reference is to the ancient Norse god of trickery, Loki. The top three feet (1 m) of a vent chimney almost 40 feet (12 m) tall at Loki's Castle in mid-July 2008. Visible at left is the arm of a remotely operated vehicle, reaching in to take fluid samples. Loki’s Castle Wents Loki's Castle - a field of five active hydrothermal vents in the mid-Atlantic Ocean - at 73 degrees north on the Mid-Atlantic Ridge - at a depth of 2,352 meters In Norse mythology, Loki is a cunning trickster who has the ability to change his shape and sex. Loki is represented as the companion of the great gods Odin and Thor. Preliminary observations have shown the warm area around the Loki's Castle vents to be alive with diverse and apparently unique microorganisms, unlike vent communities observed elsewhere. -
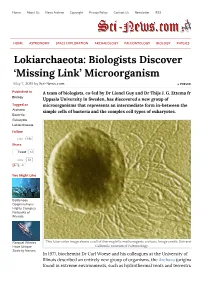
Lokiarchaeota: Biologists Discover 'Missing Link' Microorganism
Home About Us News Archive Copyright Privacy Policy Contact Us Newsletter RSS HOME ASTRONOMY SPACE EXPLORATION ARCHAEOLOGY PALEONTOLOGY BIOLOGY PHYSICS Lokiarchaeota: Biologists Discover ‘Missing Link’ Microorganism May 7, 2015 by Sci-News.com « PREVIOUS Published in A team of biologists, co-led by Dr Lionel Guy and Dr Thijs J. G. Ettema from Biology Uppsala University in Sweden, has discovered a new group of Tagged as microorganisms that represents an intermediate form in-between the Archaea simple cells of bacteria and the complex cell types of eukaryotes. Bacteria Eukaryote Lokiarchaeota Follow Like 16k Share Tweet 12 Like 58 41 You Might Like Bottlenose Dolphins Form Highly Complex Networks of Friends Rorqual Whales This false-color image shows a cell of thermophilic methanogenic archaea. Image credit: University of Have Unique California Museum of Paleontology. Stretchy Nerves In 1977, biochemist Dr Carl Woese and his colleagues at the University of Illinois described an entirely new group of organisms, the Archaea (originally found in extreme environments, such as hydrothermal vents and terrestrial hot springs). The scientists were studying relationships among the prokaryotes using DNA Extinction of sequences, and found that Archaea have distinct molecular characteristics World’s Largest separating them from bacteria as well as from eukaryotes. They proposed that Herbivores May Lead to Empty life can be divided into three domains: Eukaryota, Eubacteria, and Landscapes, Say Archaebacteria. Researchers Despite that archaeal cells were simple and small like bacteria, scientists found that Archaea were more closely related to organisms with complex cell types, a group collectively known as ‘eukaryotes.’ This observation has puzzled Sichuan Bush biologists for years. -

Radical Views of the Tree of Life
Environmental Microbiology (2009) doi:10.1111/j.1462-2920.2009.01895.x Genomics update Radical views of the Tree of Life Howard Ochman* been posited over the years (Embley and Martin, 2006; Department of Biochemistry and Molecular Biophysics, Poole and Penny, 2007). University of Arizona, Tucson, Arizona 85718, USA. To examine the origin of the eukaryotes, Pisani et al. (2007) applied a supertree approach to each of three The tripartite division of cellular organisms into three large overlapping data sets containing either 0, 8 or 21 domains – the Bacteria, the Archaea and the Eukarya – eukaryotic genomes, in order to monitor the effects of has become so ingrained that most of us have all but taxon sampling on the resulting branching orders. Super- forgotten (or did not even know) that the ancestry and trees are phylogenies that are assembled by merging sets relationships among these ancient lineages are far from of more limited trees, each based on a gene that is not settled. The most widely accepted view, which emerged necessarily present in all taxa. Their results resolved bac- primarily from analyses of anciently duplicated genes teria and archaea as separate groups, but favour a chi- (Gogarten et al., 1989; Iwabe et al., 1989), places Bacte- meric origin of eukaryotes, as the majority of eukaryotic ria as the basal lineage and distinct from the common genes with prokaryotic homologues derive from multiple ancestor of archaea and eukaryotes. Accordingly, these symbioses. These findings help explain how different studies frame the Archaea, despite their appellation, as genes originated and why particular genes show distinct archaic but not primordial, and the three domains are phylogenetic histories, but they do not ascertain whether each viewed as separate lineages, with archaea and the cellular lineage leading to eukaryotes – i.e. -

Spatial Separation of Ribosomes and DNA in Asgard Archaeal Cells ✉ ✉ Burak Avcı 1,2 , Jakob Brandt3, Dikla Nachmias4, Natalie Elia4, Mads Albertsen 3, Thijs J
www.nature.com/ismej BRIEF COMMUNICATION OPEN Spatial separation of ribosomes and DNA in Asgard archaeal cells ✉ ✉ Burak Avcı 1,2 , Jakob Brandt3, Dikla Nachmias4, Natalie Elia4, Mads Albertsen 3, Thijs J. G. Ettema 2, Andreas Schramm1,5 and Kasper Urup Kjeldsen1 © The Author(s) 2021 The origin of the eukaryotic cell is a major open question in biology. Asgard archaea are the closest known prokaryotic relatives of eukaryotes, and their genomes encode various eukaryotic signature proteins, indicating some elements of cellular complexity prior to the emergence of the first eukaryotic cell. Yet, microscopic evidence to demonstrate the cellular structure of uncultivated Asgard archaea in the environment is thus far lacking. We used primer-free sequencing to retrieve 715 almost full-length Loki- and Heimdallarchaeota 16S rRNA sequences and designed novel oligonucleotide probes to visualize their cells in marine sediments (Aarhus Bay, Denmark) using catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH). Super-resolution microscopy revealed 1–2 µm large, coccoid cells, sometimes occurring as aggregates. Remarkably, the DNA staining was spatially separated from ribosome-originated FISH signals by 50–280 nm. This suggests that the genomic material is condensed and spatially distinct in a particular location and could indicate compartmentalization or membrane invagination in Asgard archaeal cells. The ISME Journal; https://doi.org/10.1038/s41396-021-01098-3 INTRODUCTION cells in marine sediments [8] yet methodical limitations did not The origin of the eukaryotic cell is a major unresolved puzzle in allow to discern single cells and to draw any in-depth conclusion the history of life. -

Archaea and the Origin of Eukaryotes
REVIEWS Archaea and the origin of eukaryotes Laura Eme, Anja Spang, Jonathan Lombard, Courtney W. Stairs and Thijs J. G. Ettema Abstract | Woese and Fox’s 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor. Sister groups Two descendants that split The pioneering work by Carl Woese and colleagues In this Review, we discuss how culture- independent from the same node; the revealed that all cellular life could be divided into three genomics has transformed our understanding of descendants are each other’s major evolutionary lines (also called domains): the archaeal diversity and how this has influenced our closest relative. -

Newly Found Microbe Is Close Relative of Complex Life - BBC News
Newly found microbe is close relative of complex life - BBC News http://www.bbc.com/news/science-environment-32610177 Cookies on the BBC website We use cookies to ensure that we give you the best experience on our website. We also use cookies to ensure we show you advertising that is relevant to you. If you continue without changing your settings, we'll assume that you are happy to receive all cookies on the BBC website. However, if you would like to, you can change your cookie settings at any time. Continue Change settings Find out more News Sport Weather Shop Earth More Home Video World UK Business Tech Science Magazine Entertainment & Arts Health In PicturesMore ADVERTISEMENT Science & Environment By Paul Rincon Science editor, BBC News website 6 May 2015 Science & Environment The new group of archaea was discovered in sediments along the Arctic Mid-Ocean Ridge A newly discovered life form could help resolve one of the most contentious conundrums in modern biology. All organisms on Earth are classified as either prokaryotes, which have simple cells, or eukaryotes, which have larger, more complex cells. But the two cell types are so divergent that understanding how one evolved from the other has foxed biologists. The new microbes, reported in Nature journal, go some way to bridging that gap. They have been named Lokiarchaeota, partly after the Loki's Castle volcanic vent system lying 15km away from the site where the microbes' genetic material was isolated in cold marine sediments of the Arctic Mid-Ocean Ridge. 09.05.2015 14:32 Newly found microbe is close relative of complex life - BBC News http://www.bbc.com/news/science-environment-32610177 Domain names Prokaryotes are single-celled organisms and comprise all bacteria and archaea (a group of microbes that were once considered to be bacteria, but now form a separate domain of life). -

Evidence Supporting a Viral Origin of the Eukaryotic Nucleus
bioRxiv preprint doi: https://doi.org/10.1101/679175; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Evidence supporting a viral origin of the eukaryotic nucleus 2 3 4 Dr Philip JL Bell 5 Microbiogen Pty Ltd 6 Correspondence should be addressed to Email [email protected] 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 Keywords: Viral eukaryogenesis, nucleus, eukaryote origin, viral factory, mRNA capping, phylogeny 26 27 1 bioRxiv preprint doi: https://doi.org/10.1101/679175; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 28 Abstract 29 30 The defining feature of the eukaryotic cell is the possession of a nucleus that uncouples transcription 31 from translation. This uncoupling of transcription from translation depends on a complex process 32 employing hundreds of eukaryotic specific genes acting in concert and requires the 7- 33 methylguanylate (m7G) cap to prime eukaryotic mRNA for splicing, nuclear export, and cytoplasmic 34 translation. The origin of this complex system is currently a paradox since it is not found or needed 35 in prokaryotic cells which lack nuclei, yet it was apparently present and fully functional in the Last 36 Eukaryotic Common Ancestor (LECA). -
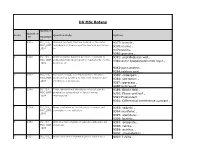
DU Msc Botany
DU MSc Botany Question Question Sr.No Question Body Options Id Descripti on 1 2345 DU_J19_ Channel proteins that are located in the outer 9377: barrels , MSC_BOT membrane of Gram-negative bacteria are known 9378:murins , _Q01 as 9379:porins, 9380:granules , 2 2346 DU_J19_ Gram-negative bacteria are more resistant to 9381: peptidoglycan wall., MSC_BOT antibiotics than Gram-positive bacteria due to the 9382:outer lipopolysaccharide layer., _Q02 presence of 9383:porin protein., 9384:teichoic acid. , 3 2347 DU_J19_ Dormant, tough, non-reproductive structure, 9385: endospore , MSC_BOT produced by bacteria to tide over unfavourable 9386: sclerotium , _Q03 conditions is known as: 9387:sporocarp , 9388:heterocyst , 4 2348 DU_J19_ Three-dimensional structures of a cell can be 9389: Bright field , MSC_BOT studied by using which of the following 9390: Phase contrast , _Q04 microscopes? 9391:Fluorescent , 9392: Differential interference contrast , 5 2349 DU_J19_ Algae that grow at the interface of water and 9393: epipelic , MSC_BOT atmosphere are called as 9394:neustonic , _Q05 9395: planktonic , 9396: benthic, 6 2350 DU_J19_ Orthorhombic crystals of calcium carbonate are 9397: aragonite., MSC_BOT known as 9398: calcite. , _Q06 9399: detritus. , 9400: stromatolites. , 7 2351 DU_J19_ Which one of the following amino acids has a 9401: Lysine , MSC_BOT nonpolar, aliphatic R group? _Q07 7 2351 DU_J19_ Which one of the following amino acids has a MSC_BOT nonpolar, aliphatic R group? 9402: Histidine , _Q07 9403: Arginine , 9404: Glycine, 8 2352 DU_J19_ -

Can We Understand Evolution Without Symbiogenesis?
Can We Understand Evolution Without Symbiogenesis? Francisco Carrapiço …symbiosis is more than a mere casual and isolated biological phenomenon: it is in reality the most fundamental and universal order or law of life. Hermann Reinheimer (1915) Abstract This work is a contribution to the literature and knowledge on evolu- tion that takes into account the biological data obtained on symbiosis and sym- biogenesis. Evolution is traditionally considered a gradual process essentially consisting of natural selection, conducted on minimal phenotypical variations that are the result of mutations and genetic recombinations to form new spe- cies. However, the biological world presents and involves symbiotic associations between different organisms to form consortia, a new structural life dimension and a symbiont-induced speciation. The acknowledgment of this reality implies a new understanding of the natural world, in which symbiogenesis plays an important role as an evolutive mechanism. Within this understanding, symbiosis is the key to the acquisition of new genomes and new metabolic capacities, driving living forms’ evolution and the establishment of biodiversity and complexity on Earth. This chapter provides information on some of the key figures and their major works on symbiosis and symbiogenesis and reinforces the importance of these concepts in our understanding of the natural world and the role they play in the establishing of the evolutionary complexity of living systems. In this context, the concept of the symbiogenic superorganism is also discussed. Keywords Evolution · Symbiogenesis · Symbiosis · Symbiogenic superorgan- ism · New paradigm F. Carrapiço (*) Centre for Ecology Evolution and Environmental Change (CE3C); Centre for Philosophy of Science, Department of Plant Biology, Faculty of science, University of Lisbon, Lisbon, Portugal e-mail: [email protected] © Springer International Publishing Switzerland 2015 81 N. -

The Gospel According to LUCA (The Last Universal Common Ancestor)
UC San Diego UC San Diego Electronic Theses and Dissertations Title The gospel according to LUCA (the last universal common ancestor) Permalink https://escholarship.org/uc/item/6pw6n8xp Author Valas, Ruben Eliezer Meyer Publication Date 2010 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The gospel according to LUCA (the last universal common ancestor) A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics by Ruben Eliezer Meyer Valas Committee in charge: Professor Philip E. Bourne, Chair Professor William F. Loomis, Co-Chair Professor Russell F. Doolittle Professor Richard D. Norris Professor Milton H. Saier Jr. 2010 The Dissertation of Ruben Eliezer Meyer Valas is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________________ ______________________________________________________________________ ______________________________________________________________________ ______________________________________________________________________ Co-chair ______________________________________________________________________ Chair University of California, San Diego 2010 iii DEDICATION I dedicate this work to Alexander “Sasha” Shulgin. Sasha is probably the wisest person I’ve met. He refuses to allow anything other than his own imagination dictates the rules of what is -

The Origin of Plastids By: Cheong Xin Chan, Ph.D
A Collaborative Learning Space for Science ABOUT FACULTY STUDENTS INTERMEDIATE CELL ORIGINS AND METABOLISM | Lead Editor: Gary Cote, Mario De Tullio The Origin of Plastids By: Cheong Xin Chan, Ph.D. & Debashish Bhattacharya, Ph.D. (Rutgers, The State University of New Jersey) © 2010 Nature Education Citation: Chan, C. X. & Bhattacharya, D. (2010) The Origin of Plastids. Nature Education 3(9):84 Plastids are core components of photosynthesis in plants and algae. Scientists are currently debating the events leading to the appearance of plastids in eukaryotic cells. Organelles, called plastids, are the main sites of photosynthesis in eukaryotic cells. Chloroplasts, as well as any other pigment containing cytoplasmic organelles that enables the harvesting and conversion of light and carbon dioxide into food and energy, are plastids. Found mainly in eukaryotic cells, plastids can be grouped into two distinctive types depending on their membrane structure: primary plastids and secondary plastids. Primary plastids are found in most algae and plants, and secondary, more-complex plastids are typically found in plankton, such as diatoms and dinoflagellates. Exploring the origin of plastids is an exciting field of research because it enhances our understanding of the basis of photosynthesis in green plants, our primary food source on planet Earth. Primary Plastids and Endosymbiosis Where did plastids originate? Their origin is explained by endosymbiosis, the act of a unicellular heterotrophic protist engulfing a free-living photosynthetic cyanobacterium and retaining it, instead of digesting it in the food vacuole (Margulis 1970; McFadden 2001; Kutschera & Niklas 2005). The captured cell (the endosymbiont) was then reduced to a functional organelle bound by two membranes, and was transmitted vertically to subsequent generations.