Fate of Organic Compounds Associated with Extractable and Bound Phases of Estuarine Sediments Deposited Under Varying Depositional Regimes
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
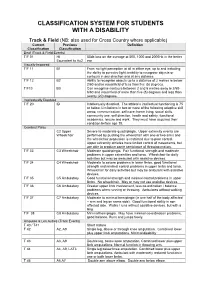
Disability Classification System
CLASSIFICATION SYSTEM FOR STUDENTS WITH A DISABILITY Track & Field (NB: also used for Cross Country where applicable) Current Previous Definition Classification Classification Deaf (Track & Field Events) T/F 01 HI 55db loss on the average at 500, 1000 and 2000Hz in the better Equivalent to Au2 ear Visually Impaired T/F 11 B1 From no light perception at all in either eye, up to and including the ability to perceive light; inability to recognise objects or contours in any direction and at any distance. T/F 12 B2 Ability to recognise objects up to a distance of 2 metres ie below 2/60 and/or visual field of less than five (5) degrees. T/F13 B3 Can recognise contours between 2 and 6 metres away ie 2/60- 6/60 and visual field of more than five (5) degrees and less than twenty (20) degrees. Intellectually Disabled T/F 20 ID Intellectually disabled. The athlete’s intellectual functioning is 75 or below. Limitations in two or more of the following adaptive skill areas; communication, self-care; home living, social skills, community use, self direction, health and safety, functional academics, leisure and work. They must have acquired their condition before age 18. Cerebral Palsy C2 Upper Severe to moderate quadriplegia. Upper extremity events are Wheelchair performed by pushing the wheelchair with one or two arms and the wheelchair propulsion is restricted due to poor control. Upper extremity athletes have limited control of movements, but are able to produce some semblance of throwing motion. T/F 33 C3 Wheelchair Moderate quadriplegia. Fair functional strength and moderate problems in upper extremities and torso. -

The Evolutionary Story Ahead of Biochemistry
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press The Organic Composition of Carbonaceous Meteorites: The Evolutionary Story Ahead of Biochemistry Sandra Pizzarello1 and Everett Shock1,2 1Department of Chemistry and Biochemistry, Arizona State University, Tempe, Arizona 85287-1604 2School of Earth and Space Exploration, Arizona State University, Tempe, Arizona 85287-1404 Correspondence: [email protected] Carbon-containing meteorites provide a natural sample of the extraterrestrial organic chemistry that occurred in the solar system ahead of life’s origin on the Earth. Analyses of 40 years have shown the organic content of these meteorites to be materials as diverse as kerogen-like macromolecules and simpler soluble compounds such as amino acids and polyols. Many meteoritic molecules have identical counterpart in the biosphere and, in a primitive group of meteorites, represent the majority of their carbon. Most of the compounds in meteorites have isotopic compositions that date their formation to presolar environments and reveal a long and active cosmochemical evolution of the biogenic elements. Whether this evolution resumed on the Earth to foster biogenesis after exogenous deliveryof meteoritic and cometary materials is not known, yet, the selective abundance of biomolecule precur- sors evident in some cosmic environments and the unique L-asymmetry of some meteoritic amino acids are suggestive of their possible contribution to terrestrial molecular evolution. INTRODUCTION that fostered biogenesis. These conditions are entirely unknown because geological and Why Meteorites are Part of the Discourse biological processes of over four billion years about the Origin of Life have long eradicated any traces of early Earth’s he studies of meteorites have long been part chemistry. -

Stabilization and Destabilization of Soil Organic Matter: Mechanisms and Controls
13F7H GEODERLIA ELSEVIER Geoderma 74 (1996) 65-105 • Stabilization and destabilization of soil organic matter: mechanisms and controls Phillip Sollins, Peter Homann, Bruce A. Caldwell Department of Forest Science Oregon State University Corvallis, OR 97331, USA Receed 1 December 1993; revised 26 July 1995; accepted 3 April 1996 Abstract We present a conceptual model of the processes by which plant leaf and root litter is transformed to soil organic C and CO 2. Stabilization of a portion of the litter C yields material that resists further transformation; destabilization yields material that is more susceptible to microbial respiration. Stability of the organic C is viewed as resulting from three general sets of characteristics. Recalcitrance comprises, molecular-level characteristics of organic substances, including elemental composition, presence of functional groups, and molecular conformation, that influence their degradation by microbes and enzymes. Interactions refers to the inter-molecular interactions between organics and either inorganic substances or other organic substances that alter the rate of degradation of those organics or synthesis of new organics. Accessibility refers to the location of organic substances with respect to microbes and enzymes. Mechanisms by which these three characteristics change through time are reviewed along with controls on those mechanisms. This review suggests that the following changes in the study of soil organic matter dynamics would speed progress: (1) increased effort to incorporate results -
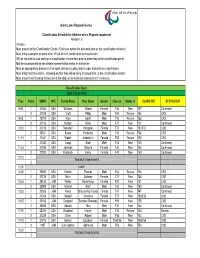
PI Classification Schedule GLRG.Xlsx
Great Lakes Regional Games Classification Schedule for Athletes with a Physical Impairment Version 1.6 Athletes - Must present to the Classification Centre 15 minutes before the allocated time on the classification schedule. Must bring a passport or some other official form of identification to classification. Will be required to read and sign a classification release form prior to presenting to the classification panel. May be accompanied by one athlete representative and/or an interpreter. Must be appropriately dressed in their sport clothes including shorts under tracksuits and sport shoes. Must bring their track chairs, strapping etc that they will be using in competition, to the classification session. Must ensure their throwing frames are at the stadium for technical assessments if necessary. Classification Day 1 Date: 9 June 2016 Time Panel SDMS NPC Family Name First Name Gender Class In Status In CLASS OUT STATUS OUT 9:00 1 31066 USA Williams Taleah Female T46 New T47 Confirmed 2 31008 USA Croft Philip Male T54 Review T54 CRS 9:45 1 15912 USA Rigo Isaiah Male T53 Review T53 CRS 2 31016 USA Nelson Brian Male F37 New F37 Confirmed 10:30 1 31218 USA Beaudoin Margaret Female T37 New T37/F37 CNS 2 30821 USA Evans Frederick Male T34 Review F34 CRS 11:15 1 11241 USA Weber Amberlynn Female T53 Review T53 CRS 2 31330 USA Langi Siale Male F43 New F43 Confirmed 11:45 1 31098 USA Johnson Shayna Female T44 New T44 Confirmed 2 27200 USA Frederick Emily Female F40 New F40 Confirmed 12:15 1 Technical Assessments 2 13:00 Lunch 14:00 1 20880 USA -

Volatile Organic Compounds, Polycyclic Aromatic Hydrocarbons and Elements in the Air of Ten Urban Homes
AIVC #13,657 J11<loor Air 2001; 11: 49-64 Copyrigfil © Mi111ksg11nl'd 2001 /itlp;//jo11mnls.1111111ksgnard.dlr/i11doornir L'IDOOR AIR Pd1lied in De1111111rk. All rights reserved ISSN 0905-6947 Volatile Organic Compounds, Polycyclic Aromatic Hydrocarbons and Elements in the Air of Ten Urban Homes MICHAEL R. VAN WINKLE1 AND PETER A. SCHEFF2* istics or occupant activities. The data indicate that several pre Abstract Ten homes were monitored at regular intervals from dietor variables, including mothball storage, air freshener use, June 1994 through April 1995 as part of a Public Health Assess and cooking activities, are reasonable predictors for emission ment in Southeast Chicago for exposure to volatile organic com rates of specific pollutants in the homes. pounds (VOCs), polycyclic aromatic hydrocarbons (PAHs), and elements. Simultaneous 24-h indoor and outdoor samples were Received for review 24 March 1999. Accepted for publication 11 March 2000. collected. VOCs were and analyzed using USEPA Method T0-14 © Indoor Air (2001) with Selected Ion Monitoring Mass Spectrometry (GC/MS). PAHs were analyzed using USEPA Method T0-13 with GC/MS. Elements were collected on quartz fiber filters and analyzed by Inductively Coupled Argon Plasma (ICP) spectroscopy or Graph ite Furnace Atomic Absorption (GFA A). Continuous measure Introduction ments of C02 and temperature were recorded for each indoor Exposure to indoor airborne pollutants is a function of sample. Twenty-four h total C02 emissions were determined from occupancy and estimated gas stove usage and were moder many variables, including pollutant infiltration, exfil 2 ately correlated (R =0.19) with 24 h average indoor C02 concen tration, deposition, resuspension, filtration, and gener trations. -

Phosphorus and Sulfur Cosmochemistry: Implications for the Origins of Life
Phosphorus and Sulfur Cosmochemistry: Implications for the Origins of Life Item Type text; Electronic Dissertation Authors Pasek, Matthew Adam Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 07/10/2021 06:16:37 Link to Item http://hdl.handle.net/10150/194288 PHOSPHORUS AND SULFUR COSMOCHEMISTRY: IMPLICATIONS FOR THE ORIGINS OF LIFE by Matthew Adam Pasek ________________________ A Dissertation Submitted to the Faculty of the DEPARTMENT OF PLANETARY SCIENCE In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY In the Graduate College UNIVERSITY OF ARIZONA 2 0 0 6 2 THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE As members of the Dissertation Committee, we certify that we have read the dissertation prepared by Matthew Adam Pasek entitled Phosphorus and Sulfur Cosmochemistry: Implications for the Origins of Life and recommend that it be accepted as fulfilling the dissertation requirement for the Degree of Doctor of Philosophy _______________________________________________________________________ Date: 04/11/2006 Dante Lauretta _______________________________________________________________________ Date: 04/11/2006 Timothy Swindle _______________________________________________________________________ Date: 04/11/2006 -

PATENT OFFICE 2,632,026 OXDATION OFAROMATIC HYDROCARBONS Joshua C
Patented Mar. 17, 1953 2,632,026 UNITED STATES PATENT OFFICE 2,632,026 OXDATION OFAROMATIC HYDROCARBONS Joshua C. Conner, Jr., Wilmington, Dei, assignor to Hercules Powder Company, Wilmington, Dei, a corporation of Delaware No Drawing. Application February 18, 1950, Serial No. 145,091 9 Cairms. (C. 260-610) 1. 2 This invention relates to a process for oxidizing of cumene at a temperature in excess of 20° C. an alkyl-substituted aromatic organic compound while simultaneously passing a stream of an having the structural formula, Oxygen-containing gas containing gaseous am Rt H monia, in catalytic amounts through the reac N / tion mixture. The reaction is continued until standard analytical data, such as refractive in R / Air dex, indicate the conversion of from about 10% to about 70% of the original cumene to oxygen in which R1 and R2 represent alkyl groups and ated products. The reaction mixture may then Ar represents either an aryl group or an alkaryl 0. be treated in accordance with known techniques group. More particularly, the invention relates to obtain a product containing preponderant to a process for the oxidation of compounds Such amounts of a,c-dimethylbenzyl hydroperoxide. as cumene in the liquid phase by naeans of no Having thus described the invention, the foll lecular. Oxygen. lowing examples are specific embodiments there It is known that cunene, for example, may be 5 of. All parts are parts by weight unless otherwise oxidized in the liquid phase by means of molec indicated. ular oxygen, but none of the processes hereto EXAMPLE 1. fore disclosed for the oxidation of cumene have resulted in substantial yields of a,c-dimethyl A glass reaction vessel equipped with a reflux benzyl hydroperoxide. -

Experimental Analysis and Modeling Aspects of the Removal of Polycyclic Aromatic Hydrocarbons in Soil Slurry Bioreactors Douglas Pino Herrera
Experimental analysis and modeling aspects of the removal of polycyclic aromatic hydrocarbons in soil slurry bioreactors Douglas Pino Herrera To cite this version: Douglas Pino Herrera. Experimental analysis and modeling aspects of the removal of polycyclic aromatic hydrocarbons in soil slurry bioreactors. Earth Sciences. Université Paris-Est, 2018. English. NNT : 2018PESC2190. tel-03012451 HAL Id: tel-03012451 https://tel.archives-ouvertes.fr/tel-03012451 Submitted on 18 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Experimental analysis and modeling aspects of the removal of PAHs in soil slurry bioreactor Douglas Oswaldo Pino Herrera Thesis committee Thesis Promotor Reviewers Prof. Mehmet A. Oturan Prof. Marie-Odile Simonnot Université Paris-Est Université de Lorraine Marne-la-Vallée, France Nancy, France Thesis Co-Promotors Prof. Francesca Beolchini Dr. Hab. Eric D. van Hullebusch, Università Politecnica delle Marche Université Paris-Est Ancona, Italy Marne-la-Vallée, France Examiners Dr. Giovanni Esposito, Prof. Piet Lens University of Cassino and Southern Lazio UNESCO-IHE Delft Cassino, Italy Delft, The Netherlands Thesis Supervisor Dr. Yannick Fayolle Dr. Yoan Pechaud Irstea Université Paris-Est Antony, France Marne-la-Vallée, France Dr. -

Athletics Classification Rules and Regulations 2
IPC ATHLETICS International Paralympic Committee Athletics Classifi cation Rules and Regulations January 2016 O cial IPC Athletics Partner www.paralympic.org/athleticswww.ipc-athletics.org @IPCAthletics ParalympicSport.TV /IPCAthletics Recognition Page IPC Athletics.indd 1 11/12/2013 10:12:43 Purpose and Organisation of these Rules ................................................................................. 4 Purpose ............................................................................................................................... 4 Organisation ........................................................................................................................ 4 1 Article One - Scope and Application .................................................................................. 6 International Classification ................................................................................................... 6 Interpretation, Commencement and Amendment ................................................................. 6 2 Article Two – Classification Personnel .............................................................................. 8 Classification Personnel ....................................................................................................... 8 Classifier Competencies, Qualifications and Responsibilities ................................................ 9 3 Article Three - Classification Panels ................................................................................ 11 4 Article Four -

Tracking Hidden Organic Carbon in Rocks Using Chemometrics And
www.nature.com/scientificreports OPEN Tracking hidden organic carbon in rocks using chemometrics and hyperspectral imaging Received: 4 October 2017 Céline Pisapia 1,2, Frédéric Jamme2, Ludovic Duponchel3 & Bénédicte Ménez1 Accepted: 25 January 2018 Finding traces of life or organic components of prebiotic interest in the rock record is an appealing goal Published: xx xx xxxx for numerous felds in Earth and space sciences. However, this is often hampered by the scarceness and highly heterogeneous distribution of organic compounds within rocks. We assess here an innovative analytical strategy combining Synchrotron radiation-based Fourier-Transform Infrared microspectroscopy (S-FTIR) and multivariate analysis techniques to track and characterize organic compounds at the pore level in complex oceanic rocks. S-FTIR hyperspectral images are analysed individually or as multiple image combinations (multiset analysis) using Principal Component Analyses (PCA) and Multivariate Curve Resolution – Alternating Least Squares (MCR-ALS). This approach allows extracting simultaneously pure organic and mineral spectral signatures and determining their spatial distributions and relationships. MCR-ALS analysis provides resolved S-FTIR signatures of 8 pure mineral and organic components showing the close association at a micrometric scale of organic compounds and secondary clays formed during rock alteration and known to catalyse organic synthesis. These results highlights the potential of the serpentinizing oceanic lithosphere to generate and preserve organic compounds of abiotic origin, in favour of the hydrothermal theory for the origin of life. Spectroscopic techniques and especially Synchrotron-based Fourier-Transform Infrared microspectroscopy (S-FTIR) have been used in geosciences for decades1–3. S-FTIR has notably been proven efcient for determining at the micrometer scale the mineral, fuid and organic phases contained in interplanetary dust particles, meteor- ites and terrestrial material4–10. -

INVESTIGATION of POLYCYCLIC AROMATIC HYDROCARBONS (Pahs) on DRY FLUE GAS DESULFURIZATION (FGD) BY-PRODUCTS
INVESTIGATION OF POLYCYCLIC AROMATIC HYDROCARBONS (PAHs) ON DRY FLUE GAS DESULFURIZATION (FGD) BY-PRODUCTS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Ping Sun, M.S. ***** The Ohio State University 2004 Dissertation Committee: Approved by Professor Linda Weavers, Adviser Professor Harold Walker Professor Patrick Hatcher Adviser Professor Yu-Ping Chin Civil Engineering Graduate Program ABSTRACT The primary goal of this research was to examine polycyclic aromatic hydrocarbons (PAHs) on dry FGD by-products to determine environmentally safe reuse options of this material. Due to the lack of information on the analytical procedures for measuring PAHs on FGD by-products, our initial work focused on analytical method development. Comparison of the traditional Soxhlet extraction, automatic Soxhlet extraction, and ultrasonic extraction was conducted to optimize the extraction of PAHs from lime spray dryer (LSD) ash (a common dry FGD by-product). Due to the short extraction time, ultrasonic extraction was further optimized by testing different organic solvents. Ultrasonic extraction with toluene as the solvent turned out to be a fast and efficient method to extract PAHs from LSD ash. The possible reactions of PAHs under standard ultrasonic extraction conditions were then studied to address concern over the possible degradation of PAHs by ultrasound. By sonicating model PAHs including naphthalene, phenanthrene and pyrene in organic solutions, extraction parameters including solvent type, solute concentration, and sonication time on reactions of PAHs were examined. A hexane: acetone (1:1 V/V) ii mixture resulted in less PAH degradation than a dichloromethane (DCM): acetone (1:1 V/V) mixture. -

Carbon Mineral Ecology: Predicting the Undiscovered Minerals of Carbon
American Mineralogist, Volume 101, pages 889–906, 2016 Carbon mineral ecology: Predicting the undiscovered minerals of carbon ROBERT M. HAZEN1,*, DANIEL R. HUMMER1, GRETHE HYSTAD2, ROBERT T. DOWNS3, AND JOSHUA J. GOLDEN3 1Geophysical Laboratory, Carnegie Institution, 5251 Broad Branch Road NW, Washington, D.C. 20015, U.S.A. 2Department of Mathematics, Computer Science, and Statistics, Purdue University Calumet, Hammond, Indiana 46323, U.S.A. 3Department of Geosciences, University of Arizona, 1040 East 4th Street, Tucson, Arizona 85721-0077, U.S.A. ABSTRACT Studies in mineral ecology exploit mineralogical databases to document diversity-distribution rela- tionships of minerals—relationships that are integral to characterizing “Earth-like” planets. As carbon is the most crucial element to life on Earth, as well as one of the defining constituents of a planet’s near-surface mineralogy, we focus here on the diversity and distribution of carbon-bearing minerals. We applied a Large Number of Rare Events (LNRE) model to the 403 known minerals of carbon, using 82 922 mineral species/locality data tabulated in http://mindat.org (as of 1 January 2015). We find that all carbon-bearing minerals, as well as subsets containing C with O, H, Ca, or Na, conform to LNRE distributions. Our model predicts that at least 548 C minerals exist on Earth today, indicating that at least 145 carbon-bearing mineral species have yet to be discovered. Furthermore, by analyzing subsets of the most common additional elements in carbon-bearing minerals (i.e., 378 C + O species; 282 C + H species; 133 C + Ca species; and 100 C + Na species), we predict that approximately 129 of these missing carbon minerals contain oxygen, 118 contain hydrogen, 52 contain calcium, and more than 60 contain sodium.