Forest Herb Pulmonaria Officinalis L
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Corynebacterium Sp.|NML98-0116
1 Limnochorda_pilosa~GCF_001544015.1@NZ_AP014924=Bacteria-Firmicutes-Limnochordia-Limnochordales-Limnochordaceae-Limnochorda-Limnochorda_pilosa 0,9635 Ammonifex_degensii|KC4~GCF_000024605.1@NC_013385=Bacteria-Firmicutes-Clostridia-Thermoanaerobacterales-Thermoanaerobacteraceae-Ammonifex-Ammonifex_degensii 0,985 Symbiobacterium_thermophilum|IAM14863~GCF_000009905.1@NC_006177=Bacteria-Firmicutes-Clostridia-Clostridiales-Symbiobacteriaceae-Symbiobacterium-Symbiobacterium_thermophilum Varibaculum_timonense~GCF_900169515.1@NZ_LT827020=Bacteria-Actinobacteria-Actinobacteria-Actinomycetales-Actinomycetaceae-Varibaculum-Varibaculum_timonense 1 Rubrobacter_aplysinae~GCF_001029505.1@NZ_LEKH01000003=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_aplysinae 0,975 Rubrobacter_xylanophilus|DSM9941~GCF_000014185.1@NC_008148=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_xylanophilus 1 Rubrobacter_radiotolerans~GCF_000661895.1@NZ_CP007514=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_radiotolerans Actinobacteria_bacterium_rbg_16_64_13~GCA_001768675.1@MELN01000053=Bacteria-Actinobacteria-unknown_class-unknown_order-unknown_family-unknown_genus-Actinobacteria_bacterium_rbg_16_64_13 1 Actinobacteria_bacterium_13_2_20cm_68_14~GCA_001914705.1@MNDB01000040=Bacteria-Actinobacteria-unknown_class-unknown_order-unknown_family-unknown_genus-Actinobacteria_bacterium_13_2_20cm_68_14 1 0,9803 Thermoleophilum_album~GCF_900108055.1@NZ_FNWJ01000001=Bacteria-Actinobacteria-Thermoleophilia-Thermoleophilales-Thermoleophilaceae-Thermoleophilum-Thermoleophilum_album -

Which Organisms Are Used for Anti-Biofouling Studies
Table S1. Semi-systematic review raw data answering: Which organisms are used for anti-biofouling studies? Antifoulant Method Organism(s) Model Bacteria Type of Biofilm Source (Y if mentioned) Detection Method composite membranes E. coli ATCC25922 Y LIVE/DEAD baclight [1] stain S. aureus ATCC255923 composite membranes E. coli ATCC25922 Y colony counting [2] S. aureus RSKK 1009 graphene oxide Saccharomycetes colony counting [3] methyl p-hydroxybenzoate L. monocytogenes [4] potassium sorbate P. putida Y. enterocolitica A. hydrophila composite membranes E. coli Y FESEM [5] (unspecified/unique sample type) S. aureus (unspecified/unique sample type) K. pneumonia ATCC13883 P. aeruginosa BAA-1744 composite membranes E. coli Y SEM [6] (unspecified/unique sample type) S. aureus (unspecified/unique sample type) graphene oxide E. coli ATCC25922 Y colony counting [7] S. aureus ATCC9144 P. aeruginosa ATCCPAO1 composite membranes E. coli Y measuring flux [8] (unspecified/unique sample type) graphene oxide E. coli Y colony counting [9] (unspecified/unique SEM sample type) LIVE/DEAD baclight S. aureus stain (unspecified/unique sample type) modified membrane P. aeruginosa P60 Y DAPI [10] Bacillus sp. G-84 LIVE/DEAD baclight stain bacteriophages E. coli (K12) Y measuring flux [11] ATCC11303-B4 quorum quenching P. aeruginosa KCTC LIVE/DEAD baclight [12] 2513 stain modified membrane E. coli colony counting [13] (unspecified/unique colony counting sample type) measuring flux S. aureus (unspecified/unique sample type) modified membrane E. coli BW26437 Y measuring flux [14] graphene oxide Klebsiella colony counting [15] (unspecified/unique sample type) P. aeruginosa (unspecified/unique sample type) graphene oxide P. aeruginosa measuring flux [16] (unspecified/unique sample type) composite membranes E. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Roseovarius Azorensis Sp. Nov., Isolated from Seawater At
Author version: Antonie van Leeuwenhoek, vol.105(3); 2014; 571-578 Roseovarius azorensis sp. nov., isolated from seawater at Espalamaca, Azores Raju Rajasabapathy • Chellandi Mohandass • Syed Gulam Dastager • Qing Liu • Thi-Nhan Khieu • Chu Ky Son • Wen-Jun Li • Ana Colaco Raju Rajasabapathy · Chellandi Mohandass* Biological Oceanography Division, CSIR-National Institute of Oceanography, Dona Paula, Goa 403 004, India. E-mail: [email protected] Syed Gulam Dastager NCIM Resource Center, CSIR-National Chemical Laboratory, Dr. Homi Bhabha road, Pune 411 008, India Qing Liu · Thi-Nhan Khieu · Wen-Jun Li Yunnan Institute of Microbiology, Yunnan University, Kunming, Yunnan 650091, P.R. China Thi-Nhan Khieu · Chu Ky Son School of Biotechnology and Food Technology, Hanoi University of Science and Technology, Vietnam Ana Colaco IMAR-Department of Oceanography and Fisheries, University Açores, Cais de Sta Cruz, 9901-862, Horta, Portugal Abstract A Gram-negative, motile, non-spore forming, rod shaped aerobic bacterium, designated strain SSW084T, was isolated from a surface seawater sample collected at Espalamaca (38°33’N; 28°39’W), Azores. Growth was found to occur from 15 – 40 °C (optimum 30 °C), at pH 7.0 – 9.0 (optimum pH 7.0) and with 25 to 100 % seawater or 0.5 – 7.0 % NaCl in the presence of Mg2+ and Ca2+; no growth was found with NaCl alone. Colonies on seawater nutrient agar (SWNA) were observed to be punctiform, white, convex, circular, smooth, and translucent. Strain SSW084T did not grow on Zobell Marine Agar (ZMA) and tryptic soy agar (TSA) even when seawater supplemented. The major respiratory quinone was found to be Q-10 and the G+C content was determined to be 61.9 mol%. -

Molecular Profiling of Culturable Bacteria from Portable Drinking Water Filtration Systems and Tap Water in Three Cities of Metro Manila, Philippines
International Journal of Philippine Science and Technology, Vol. 08, No. 2, 2015 24 ARTICLE Molecular profiling of culturable bacteria from portable drinking water filtration systems and tap water in three cities of Metro Manila, Philippines Edward A. Barlaan*, Janina M. Guarte, and Chyrene I. Moncada Molecular Diagnostics Laboratory, Institute of Biology, College of Science, University of the Philippines, Diliman, Quezon City, 1101, Philippines Abstract—Many consumers drink filtered water from portable filtration system or directly from tap water. However, microbial community composition in portable drinking water filtration systems has not yet been investigated. This study determined the molecular profile of culturable bacteria in biofilms and filtered water from portable drinking water filtration systems and tap water in three key cities of Metro Manila, Philippines. A total of 97 isolates were obtained using different growth media and characterized based on 16S rRNA gene sequences. Most bacteria were isolated from biofilms, followed by filtered water and the least from tap water. Many isolates were affiliated with Proteobacteria (α, β, and γ), Actinobacteria, Firmicutes and Bacteriodetes; some had no matches or low affiliations in data bank. Many isolates were associated with bacteria that were part of normal drinking water flora. Some were affiliated with opportunistic bacterial pathogens, soil bacteria and activated sludge bacteria. The presence of soil and opportunistic bacteria may pose health risks when immunocompromised consumers directly drink the tap water. Some isolates had very low percentage homology with bacterial affiliates or without matches in the data bank suggesting different identities or novelty of the isolates. Further studies are needed for different portable filtration systems available in the market, drinking water quality status of other areas and functions of the isolated bacteria. -

Behavioral Abnormalities of the Gut Microbiota Underlie Alzheimer’S Disease Development and Progression
Journal of Research in Medical and Dental Science 2018, Volume 6, Issue 5, Page No: 246-263 Copyright CC BY-NC 4.0 Available Online at: www.jrmds.in eISSN No. 2347-2367: pISSN No. 2347-2545 The Gut Microbiota-brain Signaling: Behavioral Abnormalities of The Gut Microbiota Underlie Alzheimer’s Disease Development and Progression. Dictatorship or Bidirectional Relationship? Menizibeya O Welcome* Department of Physiology, College of Health Sciences, The Nile University of Nigeria, Nigeria ABSTRACT Over the past decades, renewed research interest revealed crucial role of the gut microbiota in a range of health abnormalities including neurodevelopmental, neurodegenerative and neuropsychiatric diseases such as multiple sclerosis, autism spectrum disorders, and schizophrenia. More recently, emerging studies have shown that dysfunctions in gut microbiota can trigger the development or progression of Alzheimer’s disease (AD), which is the most common neurodegenerative disease worldwide. This paper presents a state-of-the-art review of recent data on the association between dysfunctions of the gut microbiota and AD development and progression. The review stresses on the functional integrity and expression of sealing and leaky junctional complexes of the intestinal and blood-brain barriers as well as contemporary understanding of the multiple mechanisms that underlie the association between barrier dysfunctions and β-amyloid accumulation, resulting to neuro inflammation and subsequently, progressive decrease in cognitive functions. Key determinants of cerebral amyloid accumulation and abnormal gut microbiota are also discussed. Very recent data on the interaction of the gut microbiota and local/distant immunocytes as well as calcium signaling defects that predispose to AD are also discussed. -

Quorum Sensing of Microalgae Associated Marine Ponticoccus Sp
Chi et al. AMB Expr (2017) 7:59 DOI 10.1186/s13568-017-0357-6 ORIGINAL ARTICLE Open Access Quorum sensing of microalgae associated marine Ponticoccus sp. PD‑2 and its algicidal function regulation Wendan Chi1, Li Zheng1,2*, Changfei He1, Bin Han1, Minggang Zheng1, Wei Gao1, Chengjun Sun1,2, Gefei Zhou3 and Xiangxing Gao4 Abstract Quorum sensing (QS) systems play important roles in regulating many physiological functions of microorganisms, such as biofilm formation, bioluminescence, and antibiotic production. One marine algicidal bacterium, Ponticoc- cus sp. PD-2, was isolated from the microalga Prorocentrum donghaiense, and its N-acyl-homoserine lactone (AHL)- mediated QS system was verified. In this study, we analyzed the AHLs profile of strain PD-2. Two AHLs, 3-oxo-C8-HSL and 3-oxo-C10-HSL, were detected using a biosensor overlay assay and GC–MS methods. Two complete AHL-QS systems (designated zlaI/R and zlbI/R) were identified in the genome of strain PD-2. When expressed in Escherichia coli, both zlaI and zlbI genes could each produce 3-oxo-C8-HSL and 3-oxo-C10-HSL. Algicidal activity was investigated by evaluating the inhibitory rate (IR) of microalgae growth by measuring the fluorescence of viable cells. We found that the metabolites of strain PD-2 had algicidal activity against its host P. donghaiense (IR 84.81%) and two other red tide microalgae, Phaeocystis globosa (IR 78.91%) and Alexandrium tamarense (IR 67.14%). β-cyclodextrin which binds to AHLs and inhibits the QS system reduced the algicidal activity more than 50%. This indicates that inhibiting the QS system may affect the algicidal metabolites production of strain PD-2. -

Cultivable Bacterial Diversity Along the Altitudinal Zonation and Vegetation Range of Tropical Eastern Himalaya
Cultivable bacterial diversity along the altitudinal zonation and vegetation range of tropical Eastern Himalaya Nathaniel A. Lyngwi1, Khedarani Koijam1, D. Sharma2 & S. R. Joshi1 1. Microbiology Laboratory, Department of Biotechnology & Bioinformatics North-Eastern Hill University, Shillong Meghalaya, India; [email protected], [email protected], [email protected], [email protected] 2. Research Officer, Regional Centre-NAEB, North-Eastern Hill University, Shillong, Meghalaya, India. Received 27-II-2012. Corrected 10-VIII-2012. Accepted 19-IX-2012. Abstract: The Northeastern part of India sprawls over an area of 262 379km2 in the Eastern Himalayan range. This constitutes a biodiversity hotspot with high levels of biodiversity and endemism; unfortunately, is also a poorly known area, especially on its microbial diversity. In this study, we assessed cultivable soil bacterial diversity and distribution from lowlands to highlands (34 to 3 990m.a.s.l.). Soil physico-chemical parameters and forest types across the different altitudes were characterized and correlated with bacterial distribution and diversity. Microbes from the soil samples were grown in Nutrient, Muller Hinton and Luria-Bertani agar plates and were initially characterized using biochemical methods. Parameters like dehydrogenase and urease activi- ties, temperature, moisture content, pH, carbon content, bulk density of the sampled soil were measured for each site. Representative isolates were also subjected to 16S rDNA sequence analysis. A total of 155 cultivable bacte- rial isolates were characterized which were analyzed for richness, evenness and diversity indices. The tropical and sub-tropical forests supported higher bacterial diversity compared to temperate pine, temperate conifer, and sub-alpine rhododendron forests. The 16S rRNA phylogenetic analysis revealed that Firmicutes was the most common group followed by Proteobacteria and Bacteroidetes. -

Inter-Domain Horizontal Gene Transfer of Nickel-Binding Superoxide Dismutase 2 Kevin M
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.12.426412; this version posted January 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Inter-domain Horizontal Gene Transfer of Nickel-binding Superoxide Dismutase 2 Kevin M. Sutherland1,*, Lewis M. Ward1, Chloé-Rose Colombero1, David T. Johnston1 3 4 1Department of Earth and Planetary Science, Harvard University, Cambridge, MA 02138 5 *Correspondence to KMS: [email protected] 6 7 Abstract 8 The ability of aerobic microorganisms to regulate internal and external concentrations of the 9 reactive oxygen species (ROS) superoxide directly influences the health and viability of cells. 10 Superoxide dismutases (SODs) are the primary regulatory enzymes that are used by 11 microorganisms to degrade superoxide. SOD is not one, but three separate, non-homologous 12 enzymes that perform the same function. Thus, the evolutionary history of genes encoding for 13 different SOD enzymes is one of convergent evolution, which reflects environmental selection 14 brought about by an oxygenated atmosphere, changes in metal availability, and opportunistic 15 horizontal gene transfer (HGT). In this study we examine the phylogenetic history of the protein 16 sequence encoding for the nickel-binding metalloform of the SOD enzyme (SodN). A comparison 17 of organismal and SodN protein phylogenetic trees reveals several instances of HGT, including 18 multiple inter-domain transfers of the sodN gene from the bacterial domain to the archaeal domain. -

Taxonomic Hierarchy of the Phylum Proteobacteria and Korean Indigenous Novel Proteobacteria Species
Journal of Species Research 8(2):197-214, 2019 Taxonomic hierarchy of the phylum Proteobacteria and Korean indigenous novel Proteobacteria species Chi Nam Seong1,*, Mi Sun Kim1, Joo Won Kang1 and Hee-Moon Park2 1Department of Biology, College of Life Science and Natural Resources, Sunchon National University, Suncheon 57922, Republic of Korea 2Department of Microbiology & Molecular Biology, College of Bioscience and Biotechnology, Chungnam National University, Daejeon 34134, Republic of Korea *Correspondent: [email protected] The taxonomic hierarchy of the phylum Proteobacteria was assessed, after which the isolation and classification state of Proteobacteria species with valid names for Korean indigenous isolates were studied. The hierarchical taxonomic system of the phylum Proteobacteria began in 1809 when the genus Polyangium was first reported and has been generally adopted from 2001 based on the road map of Bergey’s Manual of Systematic Bacteriology. Until February 2018, the phylum Proteobacteria consisted of eight classes, 44 orders, 120 families, and more than 1,000 genera. Proteobacteria species isolated from various environments in Korea have been reported since 1999, and 644 species have been approved as of February 2018. In this study, all novel Proteobacteria species from Korean environments were affiliated with four classes, 25 orders, 65 families, and 261 genera. A total of 304 species belonged to the class Alphaproteobacteria, 257 species to the class Gammaproteobacteria, 82 species to the class Betaproteobacteria, and one species to the class Epsilonproteobacteria. The predominant orders were Rhodobacterales, Sphingomonadales, Burkholderiales, Lysobacterales and Alteromonadales. The most diverse and greatest number of novel Proteobacteria species were isolated from marine environments. Proteobacteria species were isolated from the whole territory of Korea, with especially large numbers from the regions of Chungnam/Daejeon, Gyeonggi/Seoul/Incheon, and Jeonnam/Gwangju. -
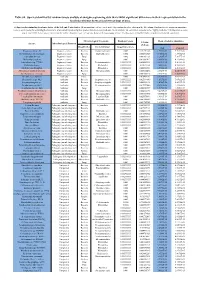
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Aestuariicoccus Marinus Gen. Nov., Sp. Nov., Isolated from Sea-Tidal Flat Sediment
TAXONOMIC DESCRIPTION Feng et al., Int J Syst Evol Microbiol 2018;68:260–265 DOI 10.1099/ijsem.0.002494 Aestuariicoccus marinus gen. nov., sp. nov., isolated from sea-tidal flat sediment Tingye Feng,1 Sang Eun Jeong,1 Kyung Hyun Kim,1 Hye Yoon Park1,2 and Che Ok Jeon1,* Abstract A Gram-stain-negative, strictly aerobic and halotolerant bacterial strain, designated strain NAP41T, was isolated from a sea tidal flat in the Yellow Sea of South Korea. Cells were non-motile cocci showing oxidase- and catalase-positive activities. Growth of strain NAP41T was observed at 15–40 C (optimum, 37 C), at pH 6.5–9.0 (optimum, pH 7.0–7.5) and in the presence of T 0.5–12 % (w/v) NaCl (optimum, 2 %). Strain NAP41 contained summed feature 8 (comprising C18 : !7c/C18 : 1!6c) and C18 : 0 as the major fatty acids and ubiquinone-10 as the sole isoprenoid quinone. Phosphatidylglycerol, phosphatidylcholine, phosphatidylethanolamine, an unidentified aminolipid and three unidentified lipids were detected as the polar lipids. The G+C content of the genomic DNA was 56.0 mol%. Strain NAP41T was most closely related to Primorskyibacter insulae SSK3-2T, Thalassococcus lentus YCS-24T and Roseivivax lentus DSM 29430T with 96.67, 96.39 and 96.39 % 16S rRNA gene sequence similarities, respectively, and formed a phylogenetic lineage distinct from closely related taxa within the family Rhodobacteraceae with low bootstrap values. On the basis of phenotypic, chemotaxonomic and molecular properties, strain NAP41T represents a novel species of a novel genus of the family Rhodobacteraceae, for which the name Aestuariicoccus marinus gen.