Command Reference for Mrbayes Ver. 3.2.5
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

COMP11 Lab 1: What the Diff?
COMP11 Lab 1: What the Diff? In this lab you will learn how to write programs that will pass our grading software with flying colors. Navigate into your COMP11 directory and download the starter code for this lab with the following command: pull-code11 lab01 (and then to see what you've got) cd lab01 ls You Do Like We Do (15 mins) There is nothing magical about how we test and grade your programs. We run your program, give it a preselected input, and check to see if its output matches our expectation. This process is repeated until we're convinced that your solution is fully functional. In this lab we will walk you through that process so that you can test your programs in the same way. Surely you recall running our encrypt demo program in Lab 0. Run this program again and specify the word \tufts" as the word to encrypt (recall that you must first enter \use comp11" if you have not added this command to your .cshrc profile). Nothing new here. But what if you wanted to do that again? What if you wanted to do it 100 more times? Having to type \tufts" during every single execution would get old fast. It would be great if you could automate the process of entering that input. Well good news: you can. It's called \redirecting". The basic concept here is simple: instead of manually entering input, you can save that input into a separate file and tell your program to read that file as if the user were entering its contents. -

1 A) Login to the System B) Use the Appropriate Command to Determine Your Login Shell C) Use the /Etc/Passwd File to Verify the Result of Step B
CSE ([email protected] II-Sem) EXP-3 1 a) Login to the system b) Use the appropriate command to determine your login shell c) Use the /etc/passwd file to verify the result of step b. d) Use the ‘who’ command and redirect the result to a file called myfile1. Use the more command to see the contents of myfile1. e) Use the date and who commands in sequence (in one line) such that the output of date will display on the screen and the output of who will be redirected to a file called myfile2. Use the more command to check the contents of myfile2. 2 a) Write a “sed” command that deletes the first character in each line in a file. b) Write a “sed” command that deletes the character before the last character in each line in a file. c) Write a “sed” command that swaps the first and second words in each line in a file. a. Log into the system When we return on the system one screen will appear. In this we have to type 100.0.0.9 then we enter into editor. It asks our details such as Login : krishnasai password: Then we get log into the commands. bphanikrishna.wordpress.com FOSS-LAB Page 1 of 10 CSE ([email protected] II-Sem) EXP-3 b. use the appropriate command to determine your login shell Syntax: $ echo $SHELL Output: $ echo $SHELL /bin/bash Description:- What is "the shell"? Shell is a program that takes your commands from the keyboard and gives them to the operating system to perform. -
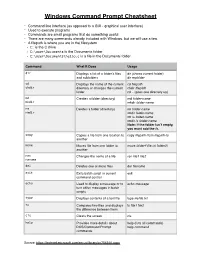
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

Don't Trust Traceroute (Completely)
Don’t Trust Traceroute (Completely) Pietro Marchetta, Valerio Persico, Ethan Katz-Bassett Antonio Pescapé University of Southern California, CA, USA University of Napoli Federico II, Italy [email protected] {pietro.marchetta,valerio.persico,pescape}@unina.it ABSTRACT In this work, we propose a methodology based on the alias resolu- tion process to demonstrate that the IP level view of the route pro- vided by traceroute may be a poor representation of the real router- level route followed by the traffic. More precisely, we show how the traceroute output can lead one to (i) inaccurately reconstruct the route by overestimating the load balancers along the paths toward the destination and (ii) erroneously infer routing changes. Categories and Subject Descriptors C.2.1 [Computer-communication networks]: Network Architec- ture and Design—Network topology (a) Traceroute reports two addresses at the 8-th hop. The common interpretation is that the 7-th hop is splitting the traffic along two Keywords different forwarding paths (case 1); another explanation is that the 8- th hop is an RFC compliant router using multiple interfaces to reply Internet topology; Traceroute; IP alias resolution; IP to Router to the source (case 2). mapping 1 1. INTRODUCTION 0.8 Operators and researchers rely on traceroute to measure routes and they assume that, if traceroute returns different IPs at a given 0.6 hop, it indicates different paths. However, this is not always the case. Although state-of-the-art implementations of traceroute al- 0.4 low to trace all the paths -

Andv's Favorite Milk Oualitv Dairv Comp Commands Bulk Tank SCC
Andv's Favorite Milk Oualitv Dairv Comp Commands Bulk Tank SCC Contribution: Command: ECON Select 6 This gives you the contribution of each cow to the bulk tank SCC. I find in most herds, the top 1% high SCC cows usually contribute less than 20o/o of the total SCC. When you see the number go to 25o/o or higher. you know that high producing cows are the main contributors. You can see the effect of removins cows from the herd on the bulk tank SCC. General Herd Information Command: Sum milk dim scc lgscc prvlg logl drylg for milk>o lact>O by lctgp This gives you a quick summary of each lactation to review. It puts all factors on one page so you can determine areas of concern. Herd SCC Status: Command: Sum lgscc=4 lgscc=6 for lgscc>O by lctgp This gives you the status of the SCC of all cows on test by lactation group and by herd. It will show you the number and percentage of cows with SCC of 200,000 and less, the number and percentage of cows with SCC of 201,000 to 800,000 and the number and percentage of cows with SCC over 800,000. My goal is to have at least 80% of all animals in the herd under 200,000. I want at least 85% of first lactation animals, 80% of second lactation animals, andl5o/o of third/older lactation animals with SCC under 200,000. New Infection Rate: Command: Sum lgscc:4 prvlg:4 for lgscc>O prvlg>O by lctgp This command only compares cows that were tested last month to the same cows tested this month. -

“Log” File in Stata
Updated July 2018 Creating a “Log” File in Stata This set of notes describes how to create a log file within the computer program Stata. It assumes that you have set Stata up on your computer (see the “Getting Started with Stata” handout), and that you have read in the set of data that you want to analyze (see the “Reading in Stata Format (.dta) Data Files” handout). A log file records all your Stata commands and output in a given session, with the exception of graphs. It is usually wise to retain a copy of the work that you’ve done on a given project, to refer to while you are writing up your findings, or later on when you are revising a paper. A log file is a separate file that has either a “.log” or “.smcl” extension. Saving the log as a .smcl file (“Stata Markup and Control Language file”) keeps the formatting from the Results window. It is recommended to save the log as a .log file. Although saving it as a .log file removes the formatting and saves the output in plain text format, it can be opened in most text editing programs. A .smcl file can only be opened in Stata. To create a log file: You may create a log file by typing log using ”filepath & filename” in the Stata Command box. On a PC: If one wanted to save a log file (.log) for a set of analyses on hard disk C:, in the folder “LOGS”, one would type log using "C:\LOGS\analysis_log.log" On a Mac: If one wanted to save a log file (.log) for a set of analyses in user1’s folder on the hard drive, in the folder “logs”, one would type log using "/Users/user1/logs/analysis_log.log" If you would like to replace an existing log file with a newer version add “replace” after the file name (Note: PC file path) log using "C:\LOGS\analysis_log.log", replace Alternately, you can use the menu: click on File, then on Log, then on Begin. -

Basic STATA Commands
Summer School on Capability and Multidimensional Poverty OPHI-HDCA, 2011 Oxford Poverty and Human Development Initiative (OPHI) http://ophi.qeh.ox.ac.uk Oxford Dept of International Development, Queen Elizabeth House, University of Oxford Basic STATA commands Typical STATA window Review Results Variables Commands Exploring your data Create a do file doedit Change your directory cd “c:\your directory” Open your database use database, clear Import from Excel (csv file) insheet using "filename.csv" Condition (after the following commands) if var1==3 or if var1==”male” Equivalence symbols: == equal; ~= not equal; != not equal; > greater than; >= greater than or equal; < less than; <= less than or equal; & and; | or. Weight [iw=weight] or [aw=weight] Browse your database browse Look for a variables lookfor “any word/topic” Summarize a variable (mean, standard su variable1 variable2 variable3 deviation, min. and max.) Tabulate a variable (per category) tab variable1 (add a second variables for cross tabs) Statistics for variables by subgroups tabstat variable1 variable2, s(n mean) by(group) Information of a variable (coding) codebook variable1, tab(99) Keep certain variables (use drop for the keep var1 var2 var3 opposite) Save a dataset save filename, [replace] Summer School on Capability and Multidimensional Poverty OPHI-HDCA, 2011 Creating Variables Generate a new variable (a number or a gen new_variable = 1 combinations of other variables) gen new_variable = variable1+ variable2 Generate a new variable conditional gen new_variable -

(PI-Net): Facial Image Obfuscation with Manipulable Semantics
Perceptual Indistinguishability-Net (PI-Net): Facial Image Obfuscation with Manipulable Semantics Jia-Wei Chen1,3 Li-Ju Chen3 Chia-Mu Yu2 Chun-Shien Lu1,3 1Institute of Information Science, Academia Sinica 2National Yang Ming Chiao Tung University 3Research Center for Information Technology Innovation, Academia Sinica Abstract Deepfakes [45], if used to replace sensitive semantics, can also mitigate privacy risks for identity disclosure [3, 15]. With the growing use of camera devices, the industry However, all of the above methods share a common has many image datasets that provide more opportunities weakness of syntactic anonymity, or say, lack of formal for collaboration between the machine learning commu- privacy guarantee. Recent studies report that obfuscated nity and industry. However, the sensitive information in faces can be re-identified through machine learning tech- the datasets discourages data owners from releasing these niques [33, 19, 35]. Even worse, the above methods are datasets. Despite recent research devoted to removing sen- not guaranteed to reach the analytical conclusions consis- sitive information from images, they provide neither mean- tent with the one derived from original images, after manip- ingful privacy-utility trade-off nor provable privacy guar- ulating semantics. To overcome the above two weaknesses, antees. In this study, with the consideration of the percep- one might resort to differential privacy (DP) [9], a rigorous tual similarity, we propose perceptual indistinguishability privacy notion with utility preservation. In particular, DP- (PI) as a formal privacy notion particularly for images. We GANs [1, 6, 23, 46] shows a promising solution for both also propose PI-Net, a privacy-preserving mechanism that the provable privacy and perceptual similarity of synthetic achieves image obfuscation with PI guarantee. -

Forest Quickstart Guide for Linguists
Forest Quickstart Guide for Linguists Guido Vanden Wyngaerd [email protected] June 28, 2020 Contents 1 Introduction 1 2 Loading Forest 2 3 Basic Usage 2 4 Adjusting node spacing 4 5 Triangles 7 6 Unlabelled nodes 9 7 Horizontal alignment of terminals 10 8 Arrows 11 9 Highlighting 14 1 Introduction Forest is a package for drawing linguistic (and other) tree diagrams de- veloped by Sašo Živanović. This manual provides a quickstart guide for linguists with just the essential things that you need to get started. More 1 extensive documentation is available from the CTAN-archive. Forest is based on the TikZ package; more information about its commands, in par- ticular those controlling the appearance of the nodes, the arrows, and the highlighting can be found in the TikZ documentation. 2 Loading Forest In your preamble, put \usepackage[linguistics]{forest} The linguistics option makes for nice trees, in which the branches meet above the two nodes that they join; it will also align the example number (provided by linguex) with the top of the tree: (1) CP C IP I VP V NP 3 Basic Usage Forest uses a familiar labelled brackets syntax. The code below will out- put the tree in (1) above (\ex. requires the linguex package and provides the example number): \ex. \begin{forest} [CP[C][IP[I][VP[V][NP]]]] \end{forest} Forest will parse the above code without problem, but you are likely to soon get lost in your labelled brackets with more complicated trees if you write the code this way. The better alternative is to arrange the nodes over multiple lines: 2 \ex. -

January 2019 Edition
Tunkhannock Area High School Tunkhannock, Pennsylvania The Prowler January 2019 Volume XIV, Issue XLVII Local Subst itute Teacher in Trouble Former TAHS substitute teacher, Zachary Migliori, faces multiple charges. By MADISON NESTOR Former substitute teacher Wyoming County Chief out, that she did not report it originally set for December at Tunkhannock Area High Detective David Ide, started to anyone. 18 was moved to March 18. School, Zachary Migliori, on October 11 when the When Detective Ide asked If he is convicted, he will was charged with three felony parents of a 15-year old Migliori if he knew that one face community service, and counts of distributing obscene student found pornographic of the girls he sent explicit mandatory counseling. material, three misdemeanor images and sexual texts messages to was a 15-year- Tunkhannock Area High counts of open lewdness, and on their daughter’s phone. old, he explained that he School took action right away three misdemeanor counts of The parent then contacted thought she was 18-years-old to ensure students’ safety, unlawful contact with minors. Detective Ide, who found because she hung out with and offers counseling to any This comes after the results after investigating that the many seniors. After being students who need it. of an investigation suspecting substitute teacher was using informed of one victim being Sources:WNEP, lewd contact with students a Snapchat account with the 15-years-old, Migliori said he WCExaminer, CitizensVoice proved to be true. According name ‘Zach Miggs.’ was disgusted with himself. to court documents, Migliori Two 17-year old females Judge Plummer set used Facebook Messenger also came forward, one of Migliori’s bail at $50,000. -

S.Ha.R.K. Installation Howto Tools Knoppix Live CD Linux Fdisk HD
S.Ha.R.K. Installation Tools HowTo • Linux fdisk utility • A copy of Linux installation CD • A copy of Windows® installation CD Tullio Facchinetti University of Pavia - Italy • Some FreeDOS utilities • A copy of S.Ha.R.K. S.Ha.R.K. Workshop S.Ha.R.K. Workshop Knoppix live CD Linux fdisk Command action a toggle a bootable flag Download ISO from b edit bsd disklabel c toggle the dos compatibility flag d delete a partition http://www.knoppix.org l list known partition types m print this menu n add a new partition o create a new empty DOS partition table p print the partition table q quit without saving changes • boot from CD s create a new empty Sun disklabel t change a partition's system id • open a command shell u change display/entry units v verify the partition table • type “su” (become root ), password is empty w write table to disk and exit x extra functionality (experts only) • start fdisk (ex. fdisk /dev/hda ) Command (m for help): S.Ha.R.K. Workshop S.Ha.R.K. Workshop HD partitioning HD partitioning 1st FreeDOS FAT32 FreeDOS must be installed Primary 2nd Windows® FAT32 into the first partition of your HD or it may not boot 3rd Linux / extX Data 1 FAT32 format data partitions as ... Extended FAT32, so that you can share Data n FAT32 your data between Linux, last Linux swap swap Windows® and FreeDOS S.Ha.R.K. Workshop S.Ha.R.K. Workshop 1 HD partitioning Windows ® installation FAT32 Windows® partition type Install Windows®.. -

What Is UNIX? the Directory Structure Basic Commands Find
What is UNIX? UNIX is an operating system like Windows on our computers. By operating system, we mean the suite of programs which make the computer work. It is a stable, multi-user, multi-tasking system for servers, desktops and laptops. The Directory Structure All the files are grouped together in the directory structure. The file-system is arranged in a hierarchical structure, like an inverted tree. The top of the hierarchy is traditionally called root (written as a slash / ) Basic commands When you first login, your current working directory is your home directory. In UNIX (.) means the current directory and (..) means the parent of the current directory. find command The find command is used to locate files on a Unix or Linux system. find will search any set of directories you specify for files that match the supplied search criteria. The syntax looks like this: find where-to-look criteria what-to-do All arguments to find are optional, and there are defaults for all parts. where-to-look defaults to . (that is, the current working directory), criteria defaults to none (that is, select all files), and what-to-do (known as the find action) defaults to ‑print (that is, display the names of found files to standard output). Examples: find . –name *.txt (finds all the files ending with txt in current directory and subdirectories) find . -mtime 1 (find all the files modified exact 1 day) find . -mtime -1 (find all the files modified less than 1 day) find . -mtime +1 (find all the files modified more than 1 day) find .