Structural Characterization of the Human Proteome
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
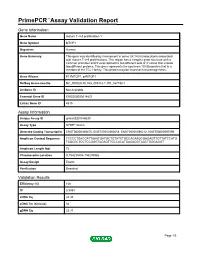
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mature T-cell proliferation 1 Gene Symbol MTCP1 Organism Human Gene Summary This gene was identified by involvement in some t(X;14) translocations associated with mature T-cell proliferations. This region has a complex gene structure with a common promoter and 5' exon spliced to two different sets of 3' exons that encode two different proteins. This gene represents the upstream 13 kDa protein that is a member of the TCL1 family. This protein may be involved in leukemogenesis. Gene Aliases P13MTCP1, p8MTCP1 RefSeq Accession No. NC_000023.10, NG_005114.1, NT_167198.1 UniGene ID Not Available Ensembl Gene ID ENSG00000214827 Entrez Gene ID 4515 Assay Information Unique Assay ID qHsaCED0048630 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369476, ENST00000362018, ENST00000596212, ENST00000597096 Amplicon Context Sequence TCCCCTGACCATTAAATGATGCTGTATCTGCCACAAGCGAGAGTTGTTATCCATG TAGCGCTCCTCCGGGTAGAGTTGCCACATGAGAGGTAGCTGGGAGGT Amplicon Length (bp) 72 Chromosome Location X:154293804-154293986 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 106 R2 0.9994 cDNA Cq 24.34 cDNA Tm (Celsius) 82 gDNA Cq 25.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report MTCP1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard -

Structural Studies of the Complex Between Akt-In and the Akt2-PH Domain Suggest That the Peptide Acts As an Allosteric Inhibitor of the Akt Kinase
The Open Spectroscopy Journal, 2009, 3, 65-76 65 Open Access Structural Studies of the Complex Between Akt-in and the Akt2-PH Domain Suggest that the Peptide Acts as an Allosteric Inhibitor of the Akt Kinase Virginie Ropars1,2,3, Philippe Barthe1,2,3, Chi-Shien Wang4, Wenlung Chen4, Der-Lii M. Tzou4,5, Anne Descours6, Loïc Martin6, Masayuki Noguchi7, Daniel Auguin1,2,3,,¶ and Christian Roumestand*,1,2,3 1CNRS UMR 5048, Centre de Biochimie Structurale, Montpellier, France 2INSERM U554, Montpellier, France 3Université Montpellier I et II, Montpellier, France 4Department of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan, ROC 5Institute of Chemistry, Academia Sinica, Nankang, Taipei 11529, Taiwan, ROC 6CEA, iBiTecs, Service d’Ingénierie Moléculaire des Protéines, 91191 Gif sur Yvette, France 7Division of Cancer Biology, Institute for Genetic Medicine, Hokkaido University, N15 W7, Kita-ku, Sapporo 060-0815, Japan Abstract: Serine/threonine kinase Akt plays a central role in the regulation of cell survival and proliferation. Hence, the search for Akt specific inhibitors constitutes an attractive strategy for anticancer therapy. We have previously demonstrated that the proto-oncogene TCL1 coactivates Akt upon binding to its Plekstrin Homology Domain, and we proposed a model for the structure of the complex TCL1:Akt2-PHD. This model led to the rational design of Akt-in, a peptide inhibitor spanning the A ß-strand of human TCL1 that binds Akt2 PH domain and inhibits the kinase activation. In the present report, we used NMR spectroscopy to determine the 3D structure of the peptide free in solution and bound to Akt2-PHD. -

Rat CLPS / Colipase Protein (His Tag)
Rat CLPS / Colipase Protein (His Tag) Catalog Number: 80729-R08H General Information SDS-PAGE: Gene Name Synonym: CLPS Protein Construction: A DNA sequence encoding the rat Clps (NP_037271.1) (Met1-Gln112) was expressed with a polyhistidine tag at the C-terminus. Source: Rat Expression Host: HEK293 Cells QC Testing Purity: > 95 % as determined by SDS-PAGE. Endotoxin: Protein Description < 1.0 EU per μg protein as determined by the LAL method. Colipase belongs to the colipase family. Structural studies of the complex Stability: and of colipase alone have revealed the functionality of its architecture. It is a small protein with five conserved disulphide bonds. Structural analogies Samples are stable for up to twelve months from date of receipt at -70 ℃ have been recognised between a developmental protein, the pancreatic lipase C-terminal domain, the N-terminal domains of lipoxygenases and the Predicted N terminal: Ala 18 C-terminal domain of alpha-toxin. Colipase can only be detected in Molecular Mass: pancreatic acinar cells, suggesting regulation of expression by tissue- specific elements. Colipase allows lipase to anchor noncovalently to the The recombinant rat Clps consists 106 amino acids and predicts a surface of lipid micelles, counteracting the destabilizing influence of molecular mass of 11.9 kDa. intestinal bile salts. Without colipase the enzyme is washed off by bile salts, which have an inhibitory effect on the lipase. Colipase is a cofactor needed Formulation: by pancreatic lipase for efficient dietary lipid hydrolysis. It binds to the C- terminal, non-catalytic domain of lipase, thereby stabilising as active Lyophilized from sterile PBS, pH 7.4. -

Biochemical Properties of Pancreatic Colipase from the Common Stingray
Ben Bacha et al. Lipids in Health and Disease 2011, 10:69 http://www.lipidworld.com/content/10/1/69 RESEARCH Open Access Biochemical properties of pancreatic colipase from the common stingray Dasyatis pastinaca Abir Ben Bacha†, Aida Karray†, Lobna Daoud, Emna Bouchaala, Madiha Bou Ali, Youssef Gargouri and Yassine Ben Ali* Background: Pancreatic colipase is a required co-factor for pancreatic lipase, being necessary for its activity during hydrolysis of dietary triglycerides in the presence of bile salts. In the intestine, colipase is cleaved from a precursor molecule, procolipase, through the action of trypsin. This cleavage yields a peptide called enterostatin knoswn, being produced in equimolar proportions to colipase. Results: In this study, colipase from the common stingray Dasyatis pastinaca (CoSPL) was purified to homogeneity. The purified colipase is not glycosylated and has an apparent molecular mass of around 10 kDa. The NH2-terminal sequencing of purified CoSPL exhibits more than 55% identity with those of mammalian, bird or marine colipases. CoSPL was found to be less effective activator of bird and mammal pancreatic lipases than for the lipase from the same specie. The apparent dissociation constant (Kd) of the colipase/lipase complex and the apparent Vmax of the colipase-activated lipase values were deduced from the linear curves of the Scatchard plots. We concluded that Stingray Pancreatic Lipase (SPL) has higher ability to interact with colipase from the same species than with the mammal or bird ones. Conclusion: The fact that colipase is a universal lipase cofactor might thus be explained by a conservation of the colipase-lipase interaction site. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Polymorphisms in PNLIP, Encoding Pancreatic Lipase, and Associations with Metabolic Traits
J Hum Genet (2001) 46:320–324 © Jpn Soc Hum Genet and Springer-Verlag 2001 ORIGINAL ARTICLE Robert A. Hegele · D. Dan Ramdath · Matthew R. Ban Michael N. Carruthers · Christine V. Carrington Henian Cao Polymorphisms in PNLIP, encoding pancreatic lipase, and associations with metabolic traits Received: January 18, 2001 / Accepted: February 19, 2001 Abstract Pancreatic lipase (EC 3.1.1.3) is an exocrine se- Introduction cretion that hydrolyzes dietary triglycerides in the small intestine. We developed genomic amplification primers to sequence the 13 exons of PNLIP, which encodes pancreatic Pancreatic lipase (PL; EC 3.1.1.3), a 56-kD protein, is in- lipase, in order to screen for possible mutations in cell lines volved in the intestinal hydrolysis of dietary triglyceride to of four children with pancreatic lipase deficiency (OMIM fatty acids. PL deficiency (OMIM 246600) is associated with 246600). We found no missense or nonsense mutations in malabsorption of long-chain triglyceride fatty acids, failure these samples, but we found three silent single-nucleotide to thrive in infancy and childhood, and absence of PL secre- polymorphisms (SNPs), namely, 96A/C in exon 3, 486C/T in tion upon secretin stimulation (Sheldon 1964; Rey et al. exon 6, and 1359C/T in exon 13. In 50 normolipidemic 1966). PNLIP on chromosome 10q26.1 (Davis et al. 1991) is Caucasians, the PNLIP 96C and 486T alleles had frequen- a 13-exon gene that spans more than 20kb (Sims et al. 1993) cies of 0.083 and 0.150, respectively. The PNLIP 1359T and encodes the 465-amino acid PL mature protein (Lowe allele was absent from Caucasian, Chinese, South Asian, et al. -

Regulation of the Akt Kinase by Interacting Proteins
Oncogene (2005) 24, 7401–7409 & 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00 www.nature.com/onc Regulation of the Akt kinase by interacting proteins Keyong Du1 and Philip N Tsichlis*,1 1Molecular Oncology Research Institute, Tufts-New England Medical Center, Boston, MA 02111, USA Ten years ago, it was observed that the Akt kinase revolutionized our understanding of cell function at is activated by phosphorylation via a phosphoinositide the molecular level.These technologies also allowed 3-kinase (PI-3K)-dependent process. This discovery gene- the identification of a large array of protein–protein rated enormous interest because it provided a link between interaction motifs that facilitate our understanding of PI-3K, an enzyme known to play a critical role in cellular signal transduction by providing clues on the potential physiology, and its downstream targets. Subsequently, it function of novel signaling proteins (Barnouin, 2004; was shown that the activity of the core components of the Miller and Stagljar, 2004). ‘PI-3K/Akt pathway’ is modulated by a complex network Akt1, also known as protein kinase Ba (PKBa) of regulatory proteins and pathways. Some of the Akt- (Bellacosa et al., 1991; Coffer and Woodgett, 1991; binding partners modulate its activation by external Jones et al., 1991), is the founding member of a protein signals by interacting with different domains of the Akt kinase family composed of three members, Akt1, Akt2, protein. This review focuses on the Akt interacting and Akt3.Akt family members regulate a diverse proteins and the mechanisms by which they regulate Akt array of cellular functions, including apoptosis, cellular activation. -

Human CLPS / Colipase Protein (His Tag)
Human CLPS / Colipase Protein (His Tag) Catalog Number: 13631-H08B General Information SDS-PAGE: Gene Name Synonym: CLPS Protein Construction: A DNA sequence encoding the human CLPS (P04118) (Met 1-Gln 112) was fused with a polyhistidine tag at the C-terminus. Source: Human Expression Host: Baculovirus-Insect Cells QC Testing Purity: > 90 % as determined by SDS-PAGE Endotoxin: Protein Description < 1.0 EU per μg of the protein as determined by the LAL method Colipase belongs to the colipase family. Structural studies of the complex Stability: and of colipase alone have revealed the functionality of its architecture. It is a small protein with five conserved disulphide bonds. Structural analogies ℃ Samples are stable for up to twelve months from date of receipt at -70 have been recognised between a developmental protein, the pancreatic lipase C-terminal domain, the N-terminal domains of lipoxygenases and the Ala 18 Predicted N terminal: C-terminal domain of alpha-toxin. Colipase can only be detected in Molecular Mass: pancreatic acinar cells, suggesting regulation of expression by tissue- specific elements. Colipase allows lipase to anchor noncovalently to the The recombinant human CLPS consists of 105 amino acids and predicts a surface of lipid micelles, counteracting the destabilizing influence of molecular mass of 11.5 kDa. It migrates as an approximately 12 KDa band in intestinal bile salts. Without colipase the enzyme is washed off by bile salts, SDS-PAGE under reducing conditions. which have an inhibitory effect on the lipase. Colipase is a cofactor needed by pancreatic lipase for efficient dietary lipid hydrolysis. It binds to the C- Formulation: terminal, non-catalytic domain of lipase, thereby stabilising as active conformation and considerably increasing the overall hydrophobic binding Lyophilized from sterile PBS, 500mM NaCl, pH 7.0, 10% gly site. -

Tcl1 Enhances Akt Kinase Activity and Mediates Its Nuclear Translocation
Tcl1 enhances Akt kinase activity and mediates its nuclear translocation Yuri Pekarsky*, Anatoliy Koval*, Cora Hallas*, Roberta Bichi*, Maria Tresini*, Scott Malstrom*, Giandomenico Russo†, Philip Tsichlis*, and Carlo M. Croce*‡ *Kimmel Cancer Center, Jefferson Medical College, Philadelphia, PA 19107; and †Istituto Dermopatico dell’Immacolata, Laboratory of Vascular Pathology, 00167 Rome, Italy Contributed by Carlo M. Croce, December 20, 1999 The TCL1 oncogene at 14q32.1 is involved in the development of wortmannin, a (PI-3K)-kinase inhibitor, completely inhibits the human mature T-cell leukemia. The mechanism of action of Tcl1 is activation of Akt (reviewed in ref. 9). Recent studies showed that unknown. Because the virus containing the v-akt oncogene causes Akt is a key player in the transduction of antiapoptotic and T-cell lymphoma in mice and Akt is a key player in transduction of proliferative signals in T-cells (refs. 10 and 11; reviewed in ref. antiapoptotic and proliferative signals in T-cells, we investigated 9). Activated Akt enhances both cell cycle progression and IL2 whether Akt and Tcl1 function in the same pathway. Coimmuno- production through the inhibition by phosphorylation of the precipitation experiments showed that endogenous Akt1 and Tcl1 proapoptotic factor Bad (11). Introduction of a constitutively physically interact in the T-cell leukemia cell line SupT11; both activated AKT1 transgene under the control of the proximal lck proteins also interact when cotransfected into 293 cells. Using promoter causes T-cell lymphomas in mice (S.M. and P.T., several AKT1 constructs in cotransfection experiments, we deter- unpublished data). In cultured cells, Akt1 can be localized in mined that this interaction occurs through the pleckstrin homology both the nucleus and cytoplasm (12). -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Uniprot Acceprotiens 121 113 Ratio(113/12 114 Ratio
Uniprot Acceprotiens 121 113 ratio(113/12 114 ratio(114/12 115 ratio(115/12 116 ratio(116/12 117 ratio(117/12 118 ratio(118/12 119 ratio(119/121) P02768 Serum albumin OS=Homo s666397.2 862466.6 1.29 593482.1 0.89 2220420.5 3.33 846469.3 1.27 634302.5 0.95 736961.1 1.11 842297.5 1.26 P02760 Protein AMBP OS=Homo s381627.7 294812.3 0.77 474165.8 1.24 203377.3 0.53 349197.6 0.92 346271.7 0.91 328356.1 0.86 411229.3 1.08 B4E1B2 cDNA FLJ53691, highly sim78511.8 107560.1 1.37 85218.8 1.09 199640.4 2.54 90022.3 1.15 73427.3 0.94 82722 1.05 102491.8 1.31 A0A0K0K1HEpididymis secretory sperm 3358.1 4584.8 1.37 4234.8 1.26 8496.1 2.53 4193.7 1.25 3507.1 1.04 3632.2 1.08 4873.3 1.45 D3DNU8 Kininogen 1, isoform CRA_302648.3 294936.6 0.97 257956.9 0.85 193831.3 0.64 290406.7 0.96 313453.3 1.04 279805.5 0.92 228883.9 0.76 B4E1C2 Kininogen 1, isoform CRA_167.9 229.7 1.37 263.2 1.57 278 1.66 326 1.94 265.5 1.58 290.3 1.73 341.5 2.03 O60494 Cubilin OS=Homo sapiens G40132.6 45037.5 1.12 38654.5 0.96 34055.8 0.85 39708.6 0.99 44702.9 1.11 45025.7 1.12 32701.3 0.81 P98164 Low-density lipoprotein rece40915.4 45344.8 1.11 35817.7 0.88 35721.8 0.87 42157.7 1.03 46693.4 1.14 48624 1.19 38847.7 0.95 A0A024RABHeparan sulfate proteoglyca46985.3 43536.1 0.93 49827.7 1.06 33964.3 0.72 44780.9 0.95 46858.6 1.00 47703.5 1.02 37785.7 0.80 P01133 Pro-epidermal growth factor 75270.8 73109.5 0.97 66336.1 0.88 56680.9 0.75 70877.8 0.94 76444.3 1.02 81110.3 1.08 65749.7 0.87 Q6N093 Putative uncharacterized pro47825.3 55632.5 1.16 48428.3 1.01 63601.5 1.33 65204.2 1.36 59384.5