The Int22h1/Int22h2-Mediated Xq28 Duplication Syndrome: an Intersection Between Neurodevelopment, Immunology, and Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
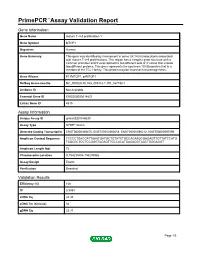
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mature T-cell proliferation 1 Gene Symbol MTCP1 Organism Human Gene Summary This gene was identified by involvement in some t(X;14) translocations associated with mature T-cell proliferations. This region has a complex gene structure with a common promoter and 5' exon spliced to two different sets of 3' exons that encode two different proteins. This gene represents the upstream 13 kDa protein that is a member of the TCL1 family. This protein may be involved in leukemogenesis. Gene Aliases P13MTCP1, p8MTCP1 RefSeq Accession No. NC_000023.10, NG_005114.1, NT_167198.1 UniGene ID Not Available Ensembl Gene ID ENSG00000214827 Entrez Gene ID 4515 Assay Information Unique Assay ID qHsaCED0048630 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369476, ENST00000362018, ENST00000596212, ENST00000597096 Amplicon Context Sequence TCCCCTGACCATTAAATGATGCTGTATCTGCCACAAGCGAGAGTTGTTATCCATG TAGCGCTCCTCCGGGTAGAGTTGCCACATGAGAGGTAGCTGGGAGGT Amplicon Length (bp) 72 Chromosome Location X:154293804-154293986 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 106 R2 0.9994 cDNA Cq 24.34 cDNA Tm (Celsius) 82 gDNA Cq 25.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report MTCP1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard -

Reframing Psychiatry for Precision Medicine
Reframing Psychiatry for Precision Medicine Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

The C9orf72-Interacting Protein Smcr8 Is a Negative Regulator of Autoimmunity and Lysosomal Exocytosis
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis Yingying Zhang,1,2,3 Aaron Burberry,1,2,3 Jin-Yuan Wang,1,2,3 Jackson Sandoe,1,2,3 Sulagna Ghosh,1,2,3 Namrata D. Udeshi,4 Tanya Svinkina,4 Daniel A. Mordes,1,2,3,5 Joanie Mok,1,2,3 Maura Charlton,1,2,3 Quan-Zhen Li,6,7 Steven A. Carr,4 and Kevin Eggan1,2,3 1Department of Stem Cell and Regenerative Biology, 2Department of Molecular and Cellular Biology, Harvard University, Cambridge, Massachusetts 02138, USA; 3Stanley Center for Psychiatric Research, Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, Massachusetts 02142, USA; 4Proteomics Platform, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA; 5Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts 02114, USA; 6Department of Immunology, 7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA While a mutation in C9ORF72 is the most common genetic contributor to amyotrophic lateral sclerosis (ALS), much remains to be learned concerning the function of the protein normally encoded at this locus. To elaborate further on functions for C9ORF72, we used quantitative mass spectrometry-based proteomics to identify interacting proteins in motor neurons and found that its long isoform complexes with and stabilizes SMCR8, which further enables interaction with WDR41. To study the organismal and cellular functions for this tripartite complex, we generated Smcr8 loss-of-function mutant mice and found that they developed phenotypes also observed in C9orf72 loss-of- function animals, including autoimmunity. -

Structural Studies of the Complex Between Akt-In and the Akt2-PH Domain Suggest That the Peptide Acts As an Allosteric Inhibitor of the Akt Kinase
The Open Spectroscopy Journal, 2009, 3, 65-76 65 Open Access Structural Studies of the Complex Between Akt-in and the Akt2-PH Domain Suggest that the Peptide Acts as an Allosteric Inhibitor of the Akt Kinase Virginie Ropars1,2,3, Philippe Barthe1,2,3, Chi-Shien Wang4, Wenlung Chen4, Der-Lii M. Tzou4,5, Anne Descours6, Loïc Martin6, Masayuki Noguchi7, Daniel Auguin1,2,3,,¶ and Christian Roumestand*,1,2,3 1CNRS UMR 5048, Centre de Biochimie Structurale, Montpellier, France 2INSERM U554, Montpellier, France 3Université Montpellier I et II, Montpellier, France 4Department of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan, ROC 5Institute of Chemistry, Academia Sinica, Nankang, Taipei 11529, Taiwan, ROC 6CEA, iBiTecs, Service d’Ingénierie Moléculaire des Protéines, 91191 Gif sur Yvette, France 7Division of Cancer Biology, Institute for Genetic Medicine, Hokkaido University, N15 W7, Kita-ku, Sapporo 060-0815, Japan Abstract: Serine/threonine kinase Akt plays a central role in the regulation of cell survival and proliferation. Hence, the search for Akt specific inhibitors constitutes an attractive strategy for anticancer therapy. We have previously demonstrated that the proto-oncogene TCL1 coactivates Akt upon binding to its Plekstrin Homology Domain, and we proposed a model for the structure of the complex TCL1:Akt2-PHD. This model led to the rational design of Akt-in, a peptide inhibitor spanning the A ß-strand of human TCL1 that binds Akt2 PH domain and inhibits the kinase activation. In the present report, we used NMR spectroscopy to determine the 3D structure of the peptide free in solution and bound to Akt2-PHD. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Autosomal Recessive Ataxias and with the Early-Onset Parkinson’S Disease That We Identified Here As Overlapping with the SFARI Autism Gene
Roadmap to Help Develop Personalized Targeted Treatments for Autism as a Disorder of the Nervous Systems Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) 1 | P a g e Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Regulation of the Akt Kinase by Interacting Proteins
Oncogene (2005) 24, 7401–7409 & 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00 www.nature.com/onc Regulation of the Akt kinase by interacting proteins Keyong Du1 and Philip N Tsichlis*,1 1Molecular Oncology Research Institute, Tufts-New England Medical Center, Boston, MA 02111, USA Ten years ago, it was observed that the Akt kinase revolutionized our understanding of cell function at is activated by phosphorylation via a phosphoinositide the molecular level.These technologies also allowed 3-kinase (PI-3K)-dependent process. This discovery gene- the identification of a large array of protein–protein rated enormous interest because it provided a link between interaction motifs that facilitate our understanding of PI-3K, an enzyme known to play a critical role in cellular signal transduction by providing clues on the potential physiology, and its downstream targets. Subsequently, it function of novel signaling proteins (Barnouin, 2004; was shown that the activity of the core components of the Miller and Stagljar, 2004). ‘PI-3K/Akt pathway’ is modulated by a complex network Akt1, also known as protein kinase Ba (PKBa) of regulatory proteins and pathways. Some of the Akt- (Bellacosa et al., 1991; Coffer and Woodgett, 1991; binding partners modulate its activation by external Jones et al., 1991), is the founding member of a protein signals by interacting with different domains of the Akt kinase family composed of three members, Akt1, Akt2, protein. This review focuses on the Akt interacting and Akt3.Akt family members regulate a diverse proteins and the mechanisms by which they regulate Akt array of cellular functions, including apoptosis, cellular activation. -

Tcl1 Enhances Akt Kinase Activity and Mediates Its Nuclear Translocation
Tcl1 enhances Akt kinase activity and mediates its nuclear translocation Yuri Pekarsky*, Anatoliy Koval*, Cora Hallas*, Roberta Bichi*, Maria Tresini*, Scott Malstrom*, Giandomenico Russo†, Philip Tsichlis*, and Carlo M. Croce*‡ *Kimmel Cancer Center, Jefferson Medical College, Philadelphia, PA 19107; and †Istituto Dermopatico dell’Immacolata, Laboratory of Vascular Pathology, 00167 Rome, Italy Contributed by Carlo M. Croce, December 20, 1999 The TCL1 oncogene at 14q32.1 is involved in the development of wortmannin, a (PI-3K)-kinase inhibitor, completely inhibits the human mature T-cell leukemia. The mechanism of action of Tcl1 is activation of Akt (reviewed in ref. 9). Recent studies showed that unknown. Because the virus containing the v-akt oncogene causes Akt is a key player in the transduction of antiapoptotic and T-cell lymphoma in mice and Akt is a key player in transduction of proliferative signals in T-cells (refs. 10 and 11; reviewed in ref. antiapoptotic and proliferative signals in T-cells, we investigated 9). Activated Akt enhances both cell cycle progression and IL2 whether Akt and Tcl1 function in the same pathway. Coimmuno- production through the inhibition by phosphorylation of the precipitation experiments showed that endogenous Akt1 and Tcl1 proapoptotic factor Bad (11). Introduction of a constitutively physically interact in the T-cell leukemia cell line SupT11; both activated AKT1 transgene under the control of the proximal lck proteins also interact when cotransfected into 293 cells. Using promoter causes T-cell lymphomas in mice (S.M. and P.T., several AKT1 constructs in cotransfection experiments, we deter- unpublished data). In cultured cells, Akt1 can be localized in mined that this interaction occurs through the pleckstrin homology both the nucleus and cytoplasm (12). -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

The Human Gene Connectome As a Map of Short Cuts for Morbid Allele Discovery
The human gene connectome as a map of short cuts for morbid allele discovery Yuval Itana,1, Shen-Ying Zhanga,b, Guillaume Vogta,b, Avinash Abhyankara, Melina Hermana, Patrick Nitschkec, Dror Friedd, Lluis Quintana-Murcie, Laurent Abela,b, and Jean-Laurent Casanovaa,b,f aSt. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; bLaboratory of Human Genetics of Infectious Diseases, Necker Branch, Paris Descartes University, Institut National de la Santé et de la Recherche Médicale U980, Necker Medical School, 75015 Paris, France; cPlateforme Bioinformatique, Université Paris Descartes, 75116 Paris, France; dDepartment of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; eUnit of Human Evolutionary Genetics, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3012, Institut Pasteur, F-75015 Paris, France; and fPediatric Immunology-Hematology Unit, Necker Hospital for Sick Children, 75015 Paris, France Edited* by Bruce Beutler, University of Texas Southwestern Medical Center, Dallas, TX, and approved February 15, 2013 (received for review October 19, 2012) High-throughput genomic data reveal thousands of gene variants to detect a single mutated gene, with the other polymorphic genes per patient, and it is often difficult to determine which of these being of less interest. This goes some way to explaining why, variants underlies disease in a given individual. However, at the despite the abundance of NGS data, the discovery of disease- population level, there may be some degree of phenotypic homo- causing alleles from such data remains somewhat limited. geneity, with alterations of specific physiological pathways under- We developed the human gene connectome (HGC) to over- come this problem. -

Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis
Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis Ugo Ala1., Rosario Michael Piro1., Elena Grassi1, Christian Damasco1, Lorenzo Silengo1, Martin Oti2, Paolo Provero1*, Ferdinando Di Cunto1* 1 Molecular Biotechnology Center, Department of Genetics, Biology and Biochemistry, University of Turin, Turin, Italy, 2 Department of Human Genetics and Centre for Molecular and Biomolecular Informatics, University Medical Centre Nijmegen, Nijmegen, The Netherlands Abstract Background: Even in the post-genomic era, the identification of candidate genes within loci associated with human genetic diseases is a very demanding task, because the critical region may typically contain hundreds of positional candidates. Since genes implicated in similar phenotypes tend to share very similar expression profiles, high throughput gene expression data may represent a very important resource to identify the best candidates for sequencing. However, so far, gene coexpression has not been used very successfully to prioritize positional candidates. Methodology/Principal Findings: We show that it is possible to reliably identify disease-relevant relationships among genes from massive microarray datasets by concentrating only on genes sharing similar expression profiles in both human and mouse. Moreover, we show systematically that the integration of human-mouse conserved coexpression with a phenotype similarity map allows the efficient identification of disease genes in large genomic regions. Finally, using this approach on 850 OMIM loci characterized by an unknown molecular basis, we propose high-probability candidates for 81 genetic diseases. Conclusion: Our results demonstrate that conserved coexpression, even at the human-mouse phylogenetic distance, represents a very strong criterion to predict disease-relevant relationships among human genes. Citation: Ala U, Piro RM, Grassi E, Damasco C, Silengo L, et al. -

Systematic Elucidation of Neuron-Astrocyte Interaction in Models of Amyotrophic Lateral Sclerosis Using Multi-Modal Integrated Bioinformatics Workflow
ARTICLE https://doi.org/10.1038/s41467-020-19177-y OPEN Systematic elucidation of neuron-astrocyte interaction in models of amyotrophic lateral sclerosis using multi-modal integrated bioinformatics workflow Vartika Mishra et al.# 1234567890():,; Cell-to-cell communications are critical determinants of pathophysiological phenotypes, but methodologies for their systematic elucidation are lacking. Herein, we propose an approach for the Systematic Elucidation and Assessment of Regulatory Cell-to-cell Interaction Net- works (SEARCHIN) to identify ligand-mediated interactions between distinct cellular com- partments. To test this approach, we selected a model of amyotrophic lateral sclerosis (ALS), in which astrocytes expressing mutant superoxide dismutase-1 (mutSOD1) kill wild-type motor neurons (MNs) by an unknown mechanism. Our integrative analysis that combines proteomics and regulatory network analysis infers the interaction between astrocyte-released amyloid precursor protein (APP) and death receptor-6 (DR6) on MNs as the top predicted ligand-receptor pair. The inferred deleterious role of APP and DR6 is confirmed in vitro in models of ALS. Moreover, the DR6 knockdown in MNs of transgenic mutSOD1 mice attenuates the ALS-like phenotype. Our results support the usefulness of integrative, systems biology approach to gain insights into complex neurobiological disease processes as in ALS and posit that the proposed methodology is not restricted to this biological context and could be used in a variety of other non-cell-autonomous communication